
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
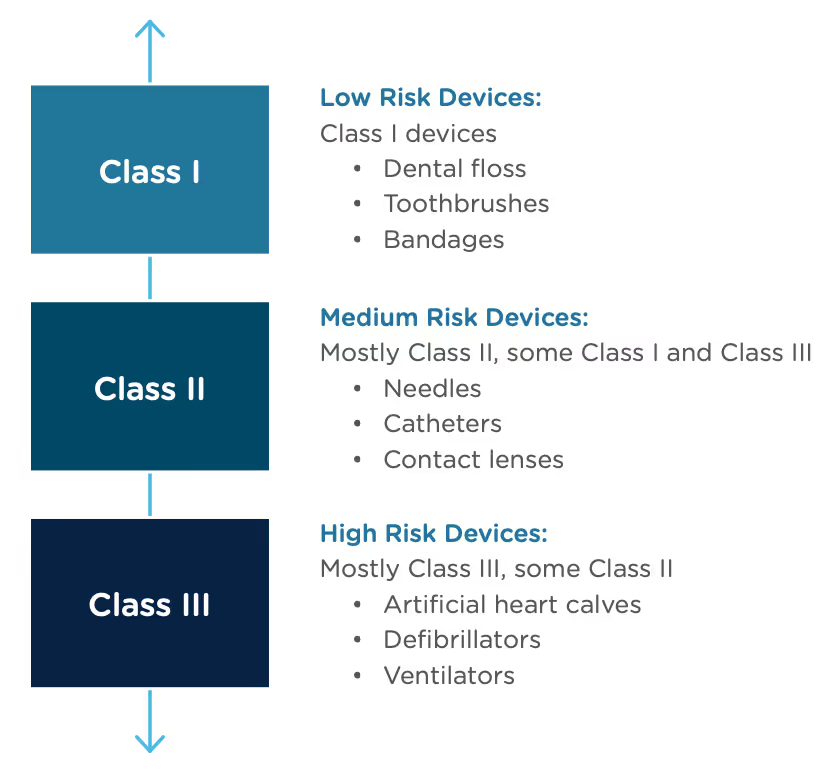
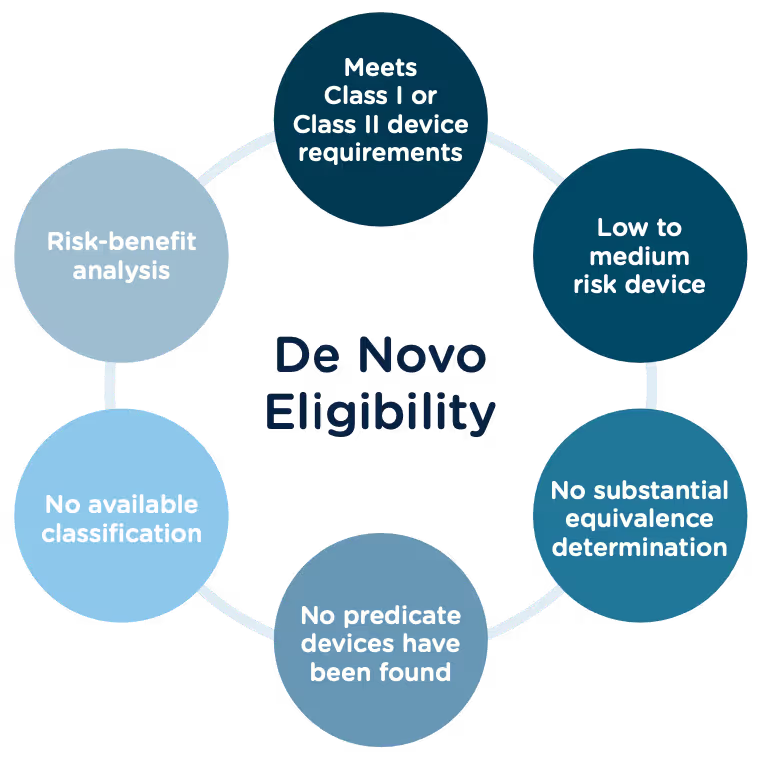
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.


GET IN TOUCH






.avif)
