
What is EU MDR?
The EU regulation 2017/745 on medical devices, or EU MDR, was a major update to medical device regulations introduced in 2017. The MDR replaces the previous EU Medical Device Directive (MDD), and is designed to modernize the EU regulatory system to better address the current needs of the market and new technologies. Devices that received a CE mark under MDD are allowed to continue to market in the EU, but will need to be recertified under MDR by a Notified Body before 2024.
The main objective of the new regulation is to strengthen protection against risks posed by medical devices and to update regulations to properly account for new technologies. Major themes of the MDR include:
- Expanded focus on regulating the entire lifecycle of a medical device
- Greater emphasis on clinical data
- Increased oversight of notified bodies
Major differences between EU MDR and MDD
The MDR is four times the size of the MDD and has an increased focus on device safety (the word safety appears 290 times in the MDR, but only 40 times in the MDD). Medical device manufacturers have found that they need to update clinical data, technical documentation, and labeling for all devices; and medical devices above class I need to be recertified by a notified body under MDR. For these reasons, companies may re-evaluate their portfolios and remove older devices from the market that don’t have adequate clinical information or yield insufficient sales to justify recertification.
Labeling (UDI and EUDAMED)
The EU MDR represents a major overhaul of medical device labeling requirements. Under the MDR, device manufacturers need to place a unique device identifier (UDI) on all devices marketed in the EU. The UDI is comprised of a UDI device identifier (‘UDI-DI’) specific to a manufacturer and a device and a UDI production identifier (‘UDI-PI’) that identifies device production characteristics. Note that there are exceptions for custom and investigational devices. In addition, UDI information must be uploaded to the new European Database on Medical Devices (EUDAMED). Together, UDI and EUDAMED are designed to allow for greater traceability and transparency of marketed devices, including improved incident reporting, field safety corrective actions, and monitoring by competent authorities. The goal is to reduce medical errors and make it more difficult for falsified devices to reach the market.
EUDAMED registration is not yet required, and changes to the specific data requirements of the database are expected. While manufacturers can enroll their device in the EUDAMED database, once that is done it must be maintained for the device. Some companies are choosing to wait until EUDAMED data requirements are finalized.
Classification rules
The MDR includes 22 classification rules, including four new rules and many updates to existing rules. Manufacturers need to verify classifications of existing devices under MDR, and may find that some devices need to be “up-classified,” resulting in more stringent regulatory requirements. Rule 11, in particular, requires the attention of any manufacturer with a device that includes software. Software that plays a part in decision-making or patient monitoring will move from a Class I to a Class IIa device. For additional information, read our recent post on Software as a Medical Device.
In addition, there are devices which were not in scope of the MDD, but are classified as medical devices under the MDR. These include products “without an intended medical purpose,” such as contact lenses.
General safety and performance requirements
MDD “Essential Requirements” have been replaced with “General Safety and Performance Requirements” in the MDR. There are 23 requirements, many of which are new, that device manufacturers will need to demonstrate conformance to. These rules place significant emphasis on risk management and “are a set of product characteristics, which are considered by the European authorities as being essential to ensuring that any new device will be safe and perform as intended throughout its life.”
Clinical evidence
New to the EU MDR is a requirement that every medical device must include sufficient clinical evidence to demonstrate compliance, dependent on the device class. This new requirement will have a significant impact on manufacturers selling existing devices without readily available clinical data.
Post-market surveillance system
MDR establishes new requirements for a post-market surveillance (PMS) system to be an integral part of the manufacturer’s Quality Management System (QMS). Post-market surveillance programs should be designed to proactively monitor safety and performance of a device, and to report any defects or issues appropriately, with all serious incidents being reported within 15 days. In addition to the many new PMS outputs, manufacturers of class IIa, IIb, and III devices are required to prepare a Periodic Safety Update Report for each device.
Person responsible for regulatory compliance (PRRC)
Under the MDR, a manufacturer needs to assign a single, qualified individual to be responsible for ensuring conformity to regulatory requirements. In addition, each Authorized Representative has to have its own PRRC.
Risk management and quality management systems
Risk management and quality controls should be in place throughout the lifecycle of the device. EN ISO 14971:2019 and EN ISO 13485:2016+A11:2021 are aligned with MDR requirements for risk management and QMS. Note that the QMS necessarily includes post-market surveillance and clinical evaluation plans.
Monitoring of notified bodies
The MDR introduced significant changes to the role and the oversight of notified bodies. The addition of post-market surveillance activities, technical documentation requirements and increased clinical requirements have placed a larger burden on the notified bodies that perform conformity assessments, which include quality system audits and technical documentation reviews. There is currently a shortage of notified bodies accredited under MDR and the industry is carefully watching for any additional extensions of MDR deadlines.
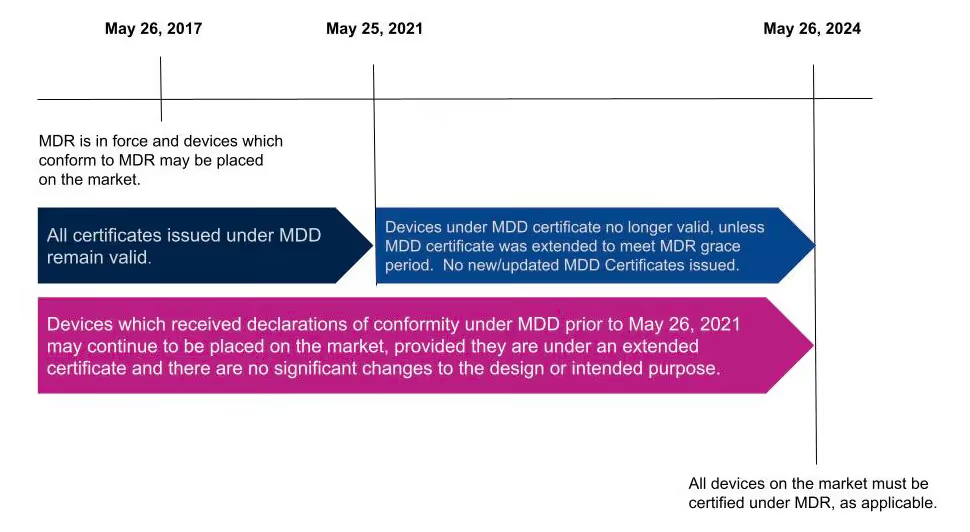
EU MDR timeline and deadlines
The MDR was published on April 5, 2017. Medical devices can currently obtain certification under MDR, but not all devices will be required to be certified under MDR until May 25, 2024.
Becoming compliant with EU MDR
Compliance with the EU MDR, EU 2017/745, requires medical device manufacturers to demonstrate that their device is designed, manufactured, and tracked according to the regulation’s requirements. Manufacturers must focus on three overall components when pursuing approval to market a medical device in the EU.
- Quality management system: A medical device must be developed with an appropriate QMS in place to ensure that a device meets its intended purpose through proper controls around design, manufacturing, and post-market surveillance.
- Clinical evidence: MDR requirements for clinical evidence are higher for most devices than in the MDD. All medical devices must demonstrate safety and efficacy for the device’s intended purpose, along with benefit-risk analysis supported by appropriate clinical evidence.
- Regulatory systems and process: The EU MDR requires more extensive processes and documentation than the MDD around quality systems, post-market surveillance tracking, risk management, on-going clinical evaluation reports, technical documentation, and more.
For more information on the EU MDR and IVDR requirements, read our Ultimate guide to the EU MDR/IVDR unique device identifier (UDI) system and Ultimate guide to the EU MDR GSPR - general safety and performance requirements.


GET IN TOUCH






.avif)
