
This article is an excerpt from The beginner's guide to the 510(k) ebook.
Table of Contents
- Introduction
- 510(k) basics
- Contents of a Traditional 510(k)
- 510(k) submission and timelines
- Other 510(k) forms
Congratulations! You have successfully developed a new medical device. Now you need to take it to market. In the United States, this often means submitting a 510(k). A 510(k) is a structured package of information about your device and its performance and safety that you submit to the Food and Drug Administration (FDA) for “clearance” before you can sell your device in the U.S. In order to receive clearance from the FDA, your 510(k) will need to demonstrate that your medical device is substantially equivalent to another legally marketed device (called a predicate device). The substantial equivalence approval process is a simple equation that looks something like this:

The 510(k) is generally the most efficient route to market clearance in the U.S. because you show your device is safe and effective based on this substantial equivalence standard, instead of needing to present more extensive clinical trial data.
There are three types of 510(k): Traditional, Abbreviated, and Special. This eBook will begin with a general overview of the 510(k) process, including its purpose and benefits. Next, we will explore the Traditional 510(k) and the sections and components required in depth. Finally, we will look at the Special and Abbreviated 510(k).
FDA: background and device oversight
Before we explain what a 510(k) is let’s first talk generally about the FDA and device oversight. The FDA is the U.S. governmental agency responsible for overseeing medical devices, drugs, food, and tobacco products. When it comes to medical devices, the FDA’s mission is to “protect the public health by ensuring the safety, efficacy, and security of…medical devices.” At the same time, the FDA also has an interest in “advancing public health by helping to speed innovations.” In other words, the FDA’s goal is to make sure devices are safe and effective for public use, while also ensuring that devices have a quick and efficient path to market.
In order to achieve this balance of safety and efficiency, the FDA has three different levels of oversight depending on the risk level of the device: (1) exempt from premarket submission, (2) Premarket Notification, also known as 510(k), and (3) Premarket Approval (PMA).
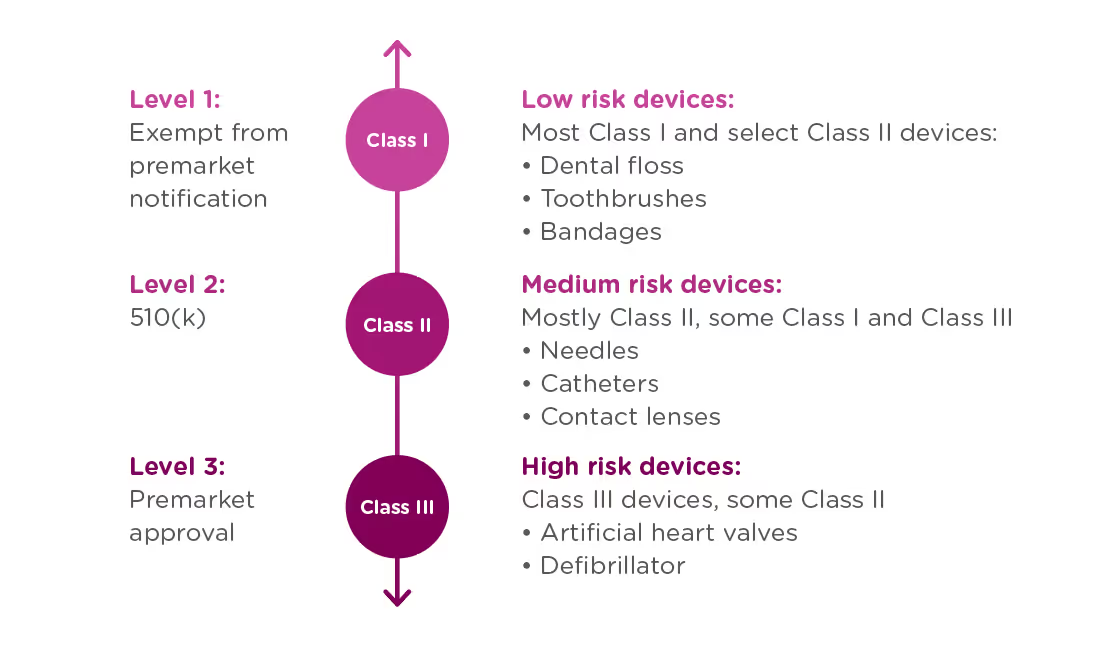
When is a 510(k) required?
A 510(k) is required for medium risk devices that have a predicate on the market which can be used to demonstrate the safety and effectiveness of the new device. Meanwhile, a PMA is required for high-risk or novel devices which require a higher level of scrutiny to be confirmed safe and effective.
A 510(k) is not only required for new devices, but also for devices that have been modified in a way that could impact safety or effectiveness. This could include changes to the:
- Design
- Components
- Materials
- Chemical composition
- Energy source
- Manufacturing process
- Intended use
You must submit your 510(k) at least 90 days before marketing the device.
What Exactly is Substantial Equivalence?
Now that we know what a 510(k) is, let’s talk about the substantial equivalence standard. You’ll recall from the introduction that your 510(k) must show that the new (or modified) device is substantially equivalent to at least one other legally marketed device, called a predicate device. Substantial equivalence looks at the intended use and the technological characteristics of the two devices.
More specifically, you must show:
- that the new device has the same intended use as the predicate, and
- the differences between the two devices do not raise questions about the safety and effectiveness of the new device.
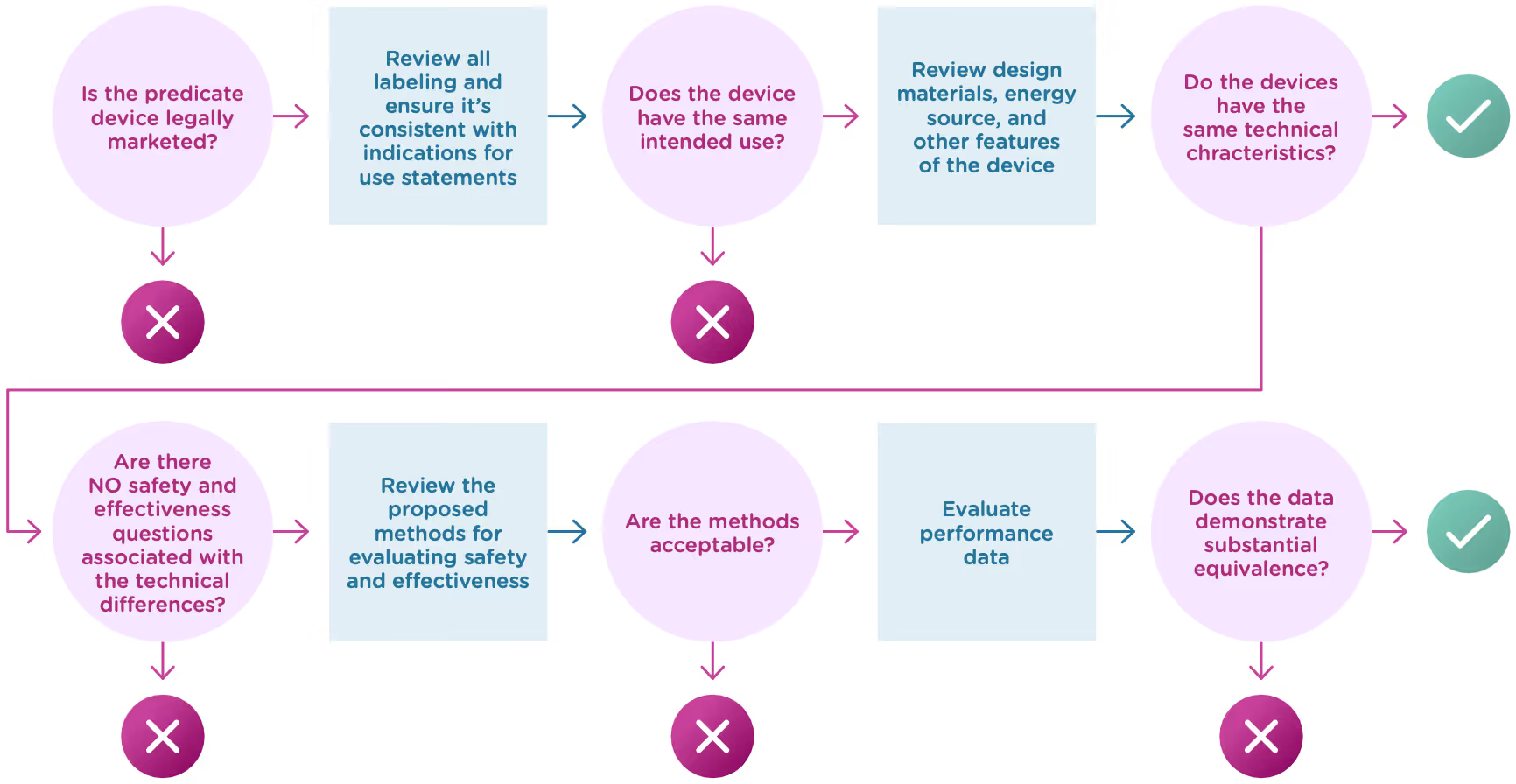
Now let’s take a closer look at intended use and technological characteristics.
Intended use
Intended use means the general purpose or function of the device. The FDA will look at your proposed labelling and your Indications of Use section of the 510(k) to determine the intended use of your device (this is covered in Chapter 2). Intended use includes:
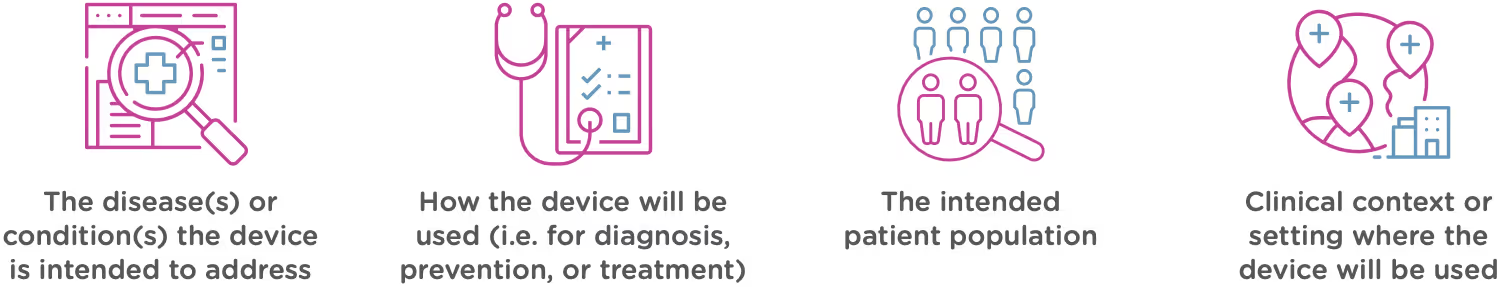
Technological characteristics
Once the FDA has determined that a predicate device exists and that the new device and the predicate device have the same intended use, it will move on to compare the technological characteristics. Technological characteristics include:
- Materials
- Design
- Energy source
- Other device features
The two devices do not have to be identical, and in fact they almost never are. The key here is to demonstrate that any differences do not have a significant impact on safety or effectiveness. Here’s what to cover when you compare your device’s technological characteristics with that of the predicate device:
Overall description of the device design
- Engineering drawings or diagrams to explain the device and component parts.
- List of component parts and explanation of how each component contributes to the overall use and function of the device.
- Physical specifications: dimensions, weight, temperature, tolerances, etc.
Materials
- Detailed chemical formulation used in all materials of constructions (especially those that come into contact with a patient).
- Any additives, coatings, paint, or surface modifications.
- How materials have been processed and what state they’re in.
Energy Sources
- Use of batteries, electricity, etc.
Other technological features
- Software/hardware
- Features
- Density
- Porosity
- Degradation characteristics
- Nature of reagents
- Principle of the assay method
In deciding whether the differences in technological characteristics impact safety or effectiveness, the FDA will typically rely on descriptive information about the technological characteristics as well as non-clinical and clinical performance data.
Let’s look at an example: A manufacturer submits a 510(k) for a new type of contact lens. Both the new device and the predicate device are indicated for daily wear for the treatment of astigmatism. The predicate device is only available in a clear lens, but the new device comes in a line of colors, including purple tinted lenses.

Who is responsible for submitting a 510(k)?
The following four types of organizations may be responsible for submitting a 510(k):
Manufacturers
- End-of-line device manufacturers who will be placing a device on the U.S. market.
- Note: Does not apply to component part manufacturers unless components will be marketed independently.
Specification developers
- Companies that develop the specifications for a finished device which has been manufactured elsewhere
Repackers or relabelers
- Required to submit a 510(k) if they significantly alter the labeling or condition of the device, including modification of manuals, changing the intended use, deleting or adding warnings, contraindications, sterilization status.
- Note: This is rare. The manufacturer, not the repackager or labeler, is typically responsible for the 510(k) submission.
Importers
- Importers that introduce a new device to the U.S. market may need to submit a 510(k), if it hasn’t already been submitted by the manufacturer.
Now that we’ve covered the basics, let’s explore what actually goes into your 510(k).
A Traditional 510(k) should contain all the following components in the list below. In some cases, a particular section may not apply to your device. When that happens, it’s a good idea to include the section anyway and just state “This section does not apply” or “N/A” under that heading.
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version


GET IN TOUCH






.avif)
