
What are medical device classes within the FDA?
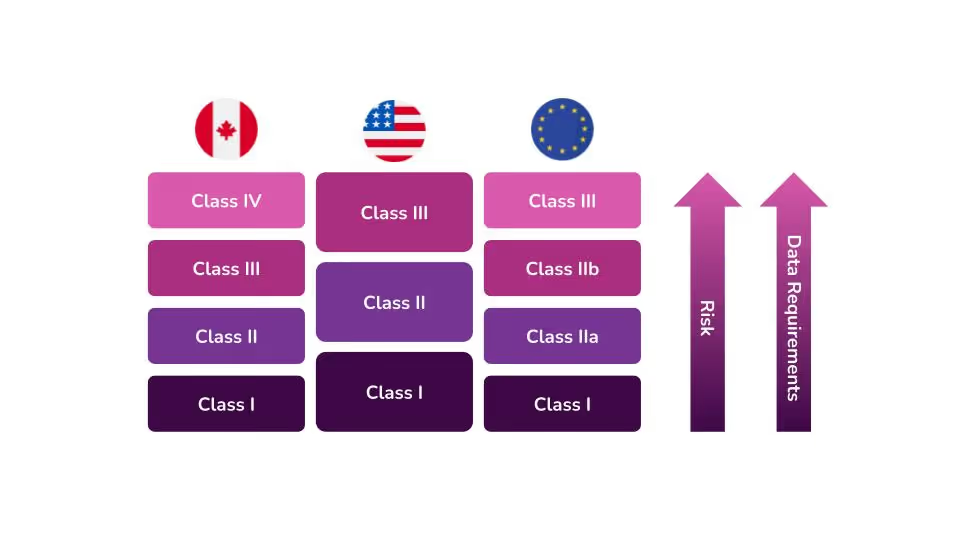
In the United States, the Food and Drug Administration (FDA) categorizes medical devices into three classes. They base these classes largely on the level of perceived risk the device may have. The level of perceived risk determines the data requirements and controls that need to be put into place to ensure safety for patients and/or users.
The FDA determines the level of oversight necessary for each device based on three factors. According to the FDA’s guidance on how to classify your device, device classification depends on the intended use of the device and also upon indications for use. The third and most important criterion is the device's risk to patients and users. The higher the risk associated with the device, the higher the class of that device will be, i.e Class I devices represent the lowest risk, and Class III devices pose the highest risk.
What are Class II Medical Devices?
Class II medical devices, which pose a medium to high risk to patients and users, account for 43 percent of all medical devices in the United States. Some common examples of Class II devices are syringes, pregnancy test kits, electric wheelchairs, and catheters. Class II medical devices must adhere to the provisions of the General Controls as mandated by the Food, Drug, and Cosmetic (FD&C) Act, which applies to all classes of devices.
The FDA also has its own Product Classification Database that can be used to assist in your device’s classification, finding consensus standards and submission types, such as a 510(k) or PMA.
What is the approval process for Class II Medical Devices?
All non-exempt Class II medical devices must go through the premarket notification process, also known as the 510(k). The 510(k) is a premarket submission process used to demonstrate that a device is safe and effective based on its substantial equivalence to a device already on the market, known as a predicate device.
Determining the substantial equivalence of your new device boils down to two things:
- Establishing that your device has the same intended use as the legally marketed predicate device
- Establishing that any technological differences between your device and the predicate device have no negative impact on the effectiveness and safety of the device
Any device with no identified predicate device, including those with a lower risk profile, is automatically classified as a Class III device and must use the more rigorous premarket approval (PMA) submission to receive market approval. Lower-risk devices can request reclassification, however, through a De Novo submission.
Class II medical devices in other countries
Device classification is different in each country. With that in mind, you should not make any assumptions regarding classification in other countries because your device is a Class II device in the United States. Each country with medical device regulations has its own classification scheme that may cause your device to be regulated differently.
During the initial phase of planning for the global commercialization of a product, it is imperative to consider international regulations, their classification schemes, and the registrations that each country will require.
The process of getting a Class II medical device to the market is arduous, and regulatory professionals must navigate disjointed manual data systems and processes. Plus, ever-evolving regulations make it difficult to ensure your new products, and even ones already on the market, are compliant.
Regulatory information management (RIM) software is an almost invaluable tool that can help your company get products to market more quickly and cost-effectively by digitizing and automating regulatory activities in a single system. The right RIM software can make the 510(k) process simpler and more efficient than ever before.
For more information on the 510(k) process, read our Beginner’s guide to the 510(k).


GET IN TOUCH






.avif)
