
What is ISO 14971?
ISO 14971 is the globally accepted international risk management standard for medical devices. This article discusses the most current version of this standard, ISO 14971:2019, currently considered the state-of-the-art standard.
ISO 14971:2019, provides the processes for identifying, evaluating, and mitigating hazards associated with the use of medical devices. While not mandatory, it is the most commonly used, industry-recognized standard to demonstrate conformity to when addressing product safety requirements. This article provides an overview of the standard, but should not be used as a substitute for the actual text of the standard.
As in the case of a quality management system, a risk management system addresses the full lifecycle of a medical device; including the design, manufacture, and use of the device. Also, while ISO 14971:2019 does not, itself, require the implementation of a quality management system, risk management is most often an important part of a strong quality management system.
Compliance with ISO 14971:2019 requires that a risk management system be established and maintained throughout the product lifecycle, and that all processes and results are stored in a risk management file. The risk management system will include processes for risk analysis, evaluation, and control. It is important to note that the standard does not define acceptable levels of risk for medical devices - this is left to the manufacturer to determine as part of their risk management processes. However, the guidance document, ISO TR 24971:2020, provides significant clarity and direction in interpreting the standard and developing a risk management system consistent with ISO 14971:2019.
EN ISO 14971:2019: EU harmonized standard
In the European Union, as of May 11, 2022, the specific version of the standard which has been officially recognized as a harmonized standard with current Medical Devices Regulation (MDR) ((EU) 2017/745 ) and In vitro Diagnostic Medical Devices Regulation (IVDR) ((EU) 2017/746), is EN ISO 14971:2019 and the amendment EN ISO 14971:2019+A11:2021. The amended version includes two Annexes, Annex ZA and ZB, which demonstrate the relationship between the standard and the risk management process required in the MDR and IVDR. The technical content of the two versions are identical and does not included any content deviations, unlike EN ISO 14971:2012, the version of the standard which is harmonized with the previous EU MDD and IVDD regulations.
Risk analysis
Under ISO 14971:2019 a manufacturer is required to document risk analysis activities and the results of those activities in a risk management file. These should include:
- Intended use and “reasonably foreseeable” misuse, along with all device characteristics which impact the safety of the device.
- Hazards (a potential source of harm*), both known and foreseeable.
- Estimation of risk for each hazard, based on the probability of occurrence of the hazard and possible consequences.
*Note: ISO 14971:2019 revises the definition of harm by excluding the word “physical” injury from the ISO 14971:2007 definition. The resulting ISO 14971:2019 definition of harm is “Injury or damage to the health of people, or damage to property or the environment”
Risk evaluation
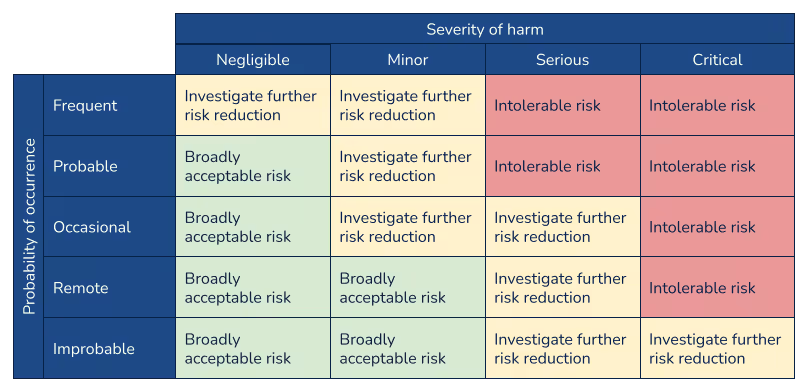
Risk evaluation involves the determination of whether a risk reduction is required for a particular hazard. Manufacturers should weigh the combination of the probability that a hazard occurs with the severity level of the hazard. A risk evaluation matrix, such as the following example, is often used to to visualize risk acceptability.
It is important to note that ISO 14971:2019 and TR 24971:2020 added significant emphasis and clarity regarding the evaluation of risk and establishment of risk acceptability criteria. Under the previous versions of the standard (both ISO 14971:2007 and EN ISO 14971:2012), there was confusion and a lack of guidance around defining acceptable risk. It was common to use a two-dimensional matrix showing severity of harm along one axis and probability of harm along the other, but with little guidance there were multiple interpretations of how to establish these criteria and these matrices were often used to define policy. The latest version of the standard and guidance, however, emphasize that the matrix should be the output of the risk management policy, which would define the criteria for risk evaluation.
Risk control
When a hazard is found to have an unacceptable risk level, risk control activities are put in place to mitigate the risk. ISO 14971:2019 requires that “state-of-the-art” best practices that are used for similar devices be employed. State-of-the-art does not necessarily mean the most advanced processes and technical features, but rather those that are generally accepted in the industry. Risk control options should include, in order of importance:
- Inherent safety by design and manufacture
- Protective measures built into the device or into the manufacturing process
- Provided safety information, and where appropriate, training to users
Risk/benefit analysis should be performed and where benefit is determined to outweigh risk, the manufacturer will need to decide what safety information is necessary to disclose.
Relevant standards should be applied as part of the risk control process whenever applicable. Some of the standards which reference ISO 14971:2019 include ISO 13485 (quality management systems), IEC 60601-1 (electrical safety), IEC/EN 62366 (usability of medical devices), and IEC 62304 (medical device software). This makes ISO 14971:2019 essential for manufacturers seeking market approval for a medical device in the U.S., European Union, Japan, Australia and many other major markets.
Production and post-production information
A substantial change in ISO 14971:2019 standard is the expansion of requirements for production and post-production activities. The manufacturer will need to perform a full review of the risk management process prior to commercial distribution. The review should ensure that the risk management plan has been appropriately implemented, the overall risk is acceptable, and that procedures are in place to gather and maintain risk data during production and post-production of the medical device. ISO 14971:2019 aligns closely with the ISO 13485:2016 section 8 requirements for feedback, analysis of data and CAPA. Information collected and reported should include any newly identified hazards, changes that affect risk analysis calculations, and results of regular reviews of the risk management file.
Management responsibilities
Medical device manufacturers who wish to demonstrate compliance with ISO 14971:2019 must have a management team that is dedicated to and supportive of the risk management system. This includes ensuring that adequate resources are assigned to support the system and that the personnel assigned are qualified for their respective responsibilities. In addition to enabling the implementation and maintenance of the risk management system, management is responsible for reviewing the system periodically to ensure continued effectiveness.
For more information about technical documentation/compliance for medical devices, check out our comprehensive ebook, The ultimate guide to EU MDR and IVDR general safety and performance requirements (GSPR).


GET IN TOUCH






.avif)
