
Medtech manufacturers cannot delay the preparation for transitioning devices to MDR. While the final deadlines were pushed, this was done primarily to address notified body capacity issues. The MDR transition period extension for legacy devices does NOT allow manufacturers to delay/deprioritize efforts until the end of the applicable extension period since many activities must be fulfilled now to utilize the extension. Read 6 reasons medtech companies shouldn’t delay MDR certification for additional information.
To illustrate the timeline, we are going to look at an example of a Class IIb non-implantable device.
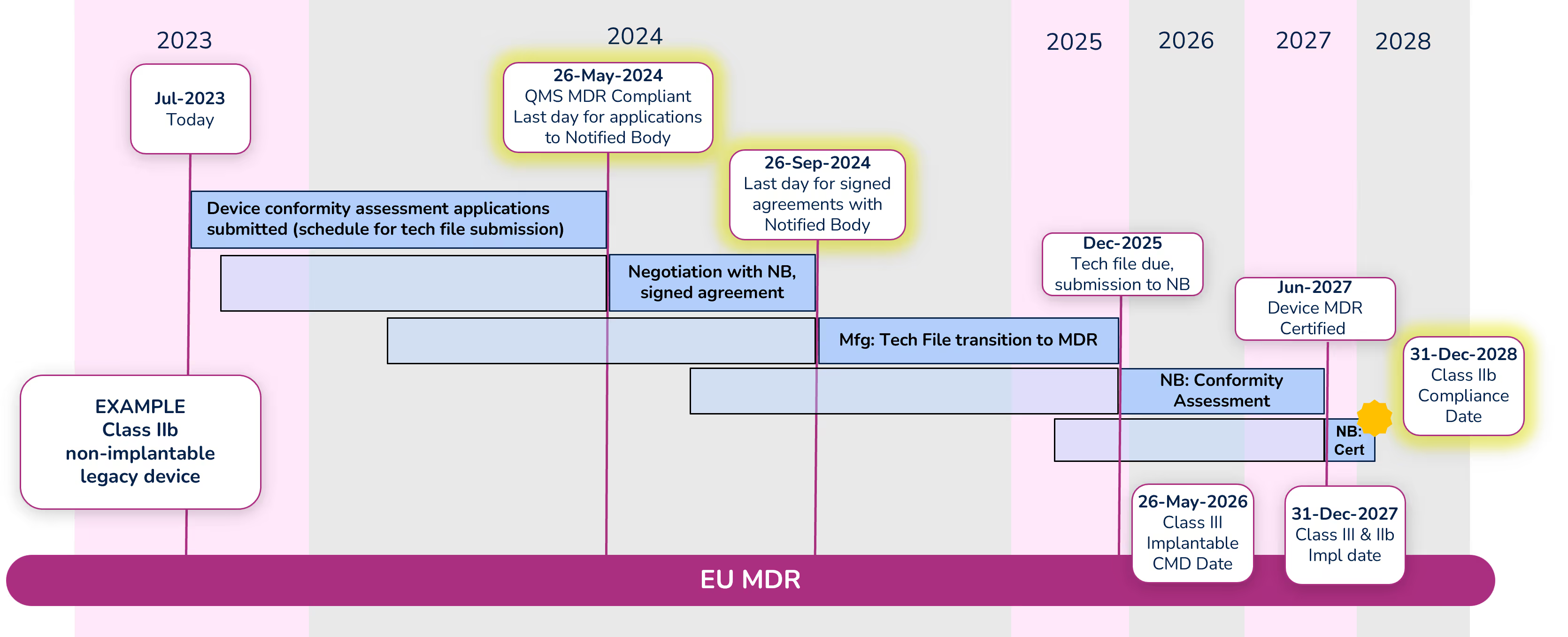
Note: Dates within the diagram for tech file submission and certification are based on your signed agreement with your Notified Body and the outcome of the conformity assessment. The dates shown are only an example.
Apply for conformity assessment - May 26, 2024
Even with the extension, a quality management system (QMS) that is compliant with MDR is required by May 26, 2024. Manufacturers must submit an application for the device conformity assessment to a Notified Body (NB) before that date as well. There are significantly fewer Notified Bodies that are certified to MDR, and due to their limited resources, manufacturers should begin working with their NB immediately if they have not already done so. Note that the device technical file does not need to be submitted with the application, but a submission schedule must be.
Sign agreement with Notified Body – September 26, 2024
Before signing an agreement with the Notified Body, expect them to counter your application with a new submission date. Manufacturers can negotiate the date with their Notified Body, but a signed agreement must be in place before September 26, 2024. While in this negotiation phase, keep in mind the MDR compliance deadline for the device (Class IIb in this example) is December 31, 2028, and work backward accounting for the various activities that must take place and their durations. It is extremely important to define a realistic schedule because not meeting that schedule will cause significant administrative complications and raise the potential risk that a device is not certified before the required deadline.
Conformity Assessment
The Notified Body will begin the conformity assessment once they receive the technical file for your device. Technical files should be submitted to the Notified Body by the date defined in your agreement (Dec 2025 in our example).
We are hearing that conformity assessments for many devices are taking 12-18 months. During that period, be prepared to answer questions and participate in conversations with your NB. In some cases, there can be significant back-and-forth between the manufacturer and the NB during the conformity assessment. Once the conformity assessment is complete (Jun 2027 in our example) it can take up to another 3-6 months for certificate issuance.
MDR extension reference documents
- Q&A Document for Regulation 2023/607 – Published by the EU Commission, this document answers some of the common questions around the extension in “plain English.”
- Regulation 2023/607 amending MDR (EU) 2017/745 and IVDR (EU) 2017/746 in regard to the transitional provisions and removal of sell-off periods for medical devices and for in vitro diagnostic medical devices.
- Notified Body Confirmation Letter template published by Team NB. This is a confirmation letter created by the manufacturer for the Notified Body to sign. The letter is not mandatory but is strongly recommended to provide objective evidence that the conditions of the extension have been met extending the validity of the CE certificate.
For more information, watch the replay of our recent "Ask Us Anything" webinar on the EU MDR Transition Period Extension.

GET IN TOUCH






.avif)
