
This article is an excerpt from Post-market surveillance for medical device in the European Union.
Table of Contents
- What is post-market surveillance?
- What classes of medical devices require post-market surveillance?
- Components of a successful post-market surveillance plan
- PMS data requirements
- Post-market surveillance system goals
- Required post-market surveillance reporting
- Embracing post-market surveillance as an integral part of your quality program
- Getting started with post-market surveillance
Post-market surveillance (PMS) is designed to monitor the performance of a marketed medical device by collecting and analyzing field use data. Article 10 of the EU MDR and IVDR requires all device manufacturers to have a post-market surveillance system in place. The main elements of the PMS are laid out in Article 83, and additional details for lower-risk and higher-risk devices are covered in articles 84 and85, respectively.
In general, a PMS system consists of both proactive activities and reactive, or vigilance, activities. While post-market surveillance and vigilance are sometimes used interchangeably, vigilance consists of separate activities that feed post-market surveillance programs.
Post-market surveillance systems are used to collect and analyze data not only about the manufacturer’s device but also about related competitors’ devices that are on the market. Data collected through PMS procedures is then used to identify trends that may lead to, among other things, quality improvements, updates to user training and instructions for use, and identification of manufacturing issues.
Note that “market surveillance” encompasses activities performed by a Competent Authority to verify MDR compliance, and should not be confused with the topic of this ebook,“post-market surveillance," which is performed by the manufacturer.
All medical devices marketed in the EU require some level of post-market surveillance, and all medical device manufacturers must implement a post-market surveillance system (PMS). The requirements of the PMS, however, vary and should be “proportionate to the risk class and appropriate for the type of device” (MDR Chapter VII). In particular, the type and frequency of reporting vary based on a device’s risk class.
A post-market surveillance plan (PMS) is an integral part of a manufacturer’s quality management system and provides a system for compiling and analyzing data that is relevant to product quality, performance, and safety throughout the entire lifetime of a device. The PMS should also provide methods for determining the need for and implementing any preventative and corrective actions. A PMS system should include and define:
Surveillance data sources
With the increased focus on proactive risk identification in the MDR, it is important to design post-market surveillance systems that actively acquire knowledge and detect potential risks. It is not sufficient to rely solely on spontaneous reporting by healthcare providers, patients, and other stakeholders.
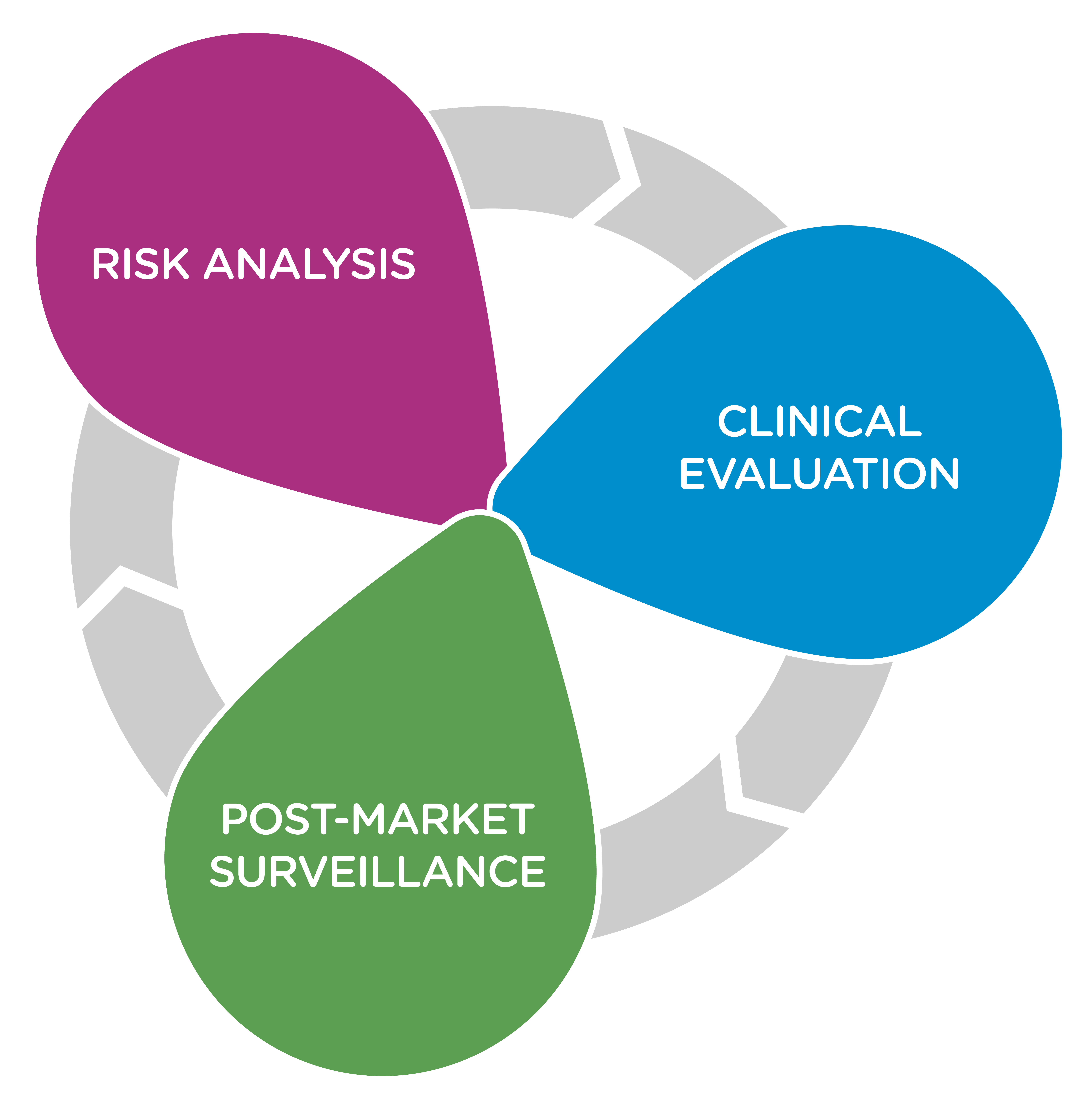
In addition to information coming from Clinical Evaluation Reports and complaint and adverse event reporting, typical sources of surveillance data include:
• Social media networks: Because many of your stakeholders may be communicating on social media networks, it is important to employ social listening techniques and/or tools to identify issues and concerning trends as they develop.
• Industry and academic literature: Any studies, academic papers, and other literature that addresses similar devices or the specific use cases for which your device is designed should be evaluated. In particular, risk factors and adverse events identified with similar devices should be closely examined. It is also important to identify newer technologies that may affect the benefit-risk ratio and establish a new definition of “state of the art” for the device type.
• EUDAMED: While the European Database on Medical Devices (EUDAMED) is not yet fully functional, it is intended to provide a living picture of the lifecycle of all medical devices marketed in the EU. Manufacturers should take special care to consider information for similar devices made available through the EUDAMED system in the future.
• Registries: Patient, disease, and device registries can provide information that informs the clinical evaluation process which provides input into the post-market surveillance system.
Data analysis methodology
A well-defined data analysis methodology will accurately identify trends and lead to defendable decisions in the application of post-market experience. Once the necessary information has been identified and collected, and potentially cleaned of incomplete or otherwise unusable data, the data needs to be analyzed.
The goal is to identify meaningful trends, correlations, variations, and patterns that can lead to improvements in the safety and efficacy of the device. There are many data analysis tools available that can assist with:
• Regression analysis that will identify correlations between data (e.g. the device location/geography correlates to battery life).
• Data visualization that can be useful in spotting trends in the data.
• Predictive analytics, which can be particularly useful with large data sets, to identify future trends based on historical data.
• Data mining, which is also normally used with large datasets, to organize data and identify data groups for further analysis.
Benefit-risk indicators and thresholds
The MDR requires that medical device manufacturers not only demonstrate the clinical benefit of their device but also quantify the benefit-risk ratio. The benefit of a device must be shown to clearly outweigh the risk for it to gain market approval. Article 2 (24) of the MDR defines the benefit-risk determination as “the analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose when used in accordance with the intended purpose given by the manufacturer.”
A PMS system should clearly define benefit-risk calculations and the data used to support them. Post-market surveillance activities are critical in order to re-evaluate and maintain the benefit-risk calculations and determinations of a device throughout its life. Information that is gained through a PMS system can lead to:
• Identification of new risk factors.
• Adjustments to risk frequency and/or severity values based on actual use data.
• Adjustments to established risk calculations based on new “state of the art” technologies becoming available.
• Adjustments to established benefit calculations based on actual use data.
While complaint handling and other feedback tracking are more often described as part of post-market vigilance systems, they play a role in the more proactive post-market surveillance processes as well. A PMS system should define ...
To continue reading this ebook, download the full version.


GET IN TOUCH






.avif)
