
This article is an excerpt from The ultimate guide to the EU MDR/IVDR UDI ebook.
Table of contents
- Overview
- UDI basics and benefits
- UDI format requirements and issuing entities
- UDI rules for specific device types
- Implementation of UDI and UDAMED in the European Union
- US vs EU UDI comparison
The EU Medical Device Regulation (2017/745) (“MDR”) and EU In Vitro Diagnosis Regulation (2017/746) (“IVDR”) introduce two new systems for information exchange: UDI (Unique Device Identifier) for device identification and EUDAMED (European Databank on Medical Devices) to centralize and disseminate information. UDI is a specific code assigned to all devices and higher levels of packaging. This will allow for devices being sold in the European market to be identified and traced through a globally harmonized approach. EUDAMED is the IT system developed by the European Commission to replace the EUDAMED2 database previously in place under the Medical Device Directives (MDD). EUDAMED is a multi-functional system that will be used to coordinate device registration, provide information about devices to industry professionals and the public, and highlight necessary safety details.
The EU MDR and IVDR UDI system is based upon the guidance of the International Medical Device Regulators Forum (IMDRF). It’s a globally harmonized system that’s designed to increase patient safety and optimize care.
UDI system goals
Increase patient safety
- Improve tracing of devices
- Reduce the presence of counterfeit devices
Ensure access to accurate information
- Unambiguous identification of devices throughout distribution and use
Improve post-market surveillance
- Improve accessibility of adverse event reports
Enhance supply chain Management
- Streamline supply chain process and inventory management
- Simplify medical device documentation processes
The UDI system has four key elements
Element 1: Assignment of UDI (UDI Components)
The first element of the UDI system is the assignment of a UDI. The UDI is a code of alphanumeric characters that acts as the access key to information about a specific medical device on the market. The EU MDR and EU IVDR requires that a UDI be assigned to all medical devices except for custom-made or investigational devices. There are three components of a UDI:
- Basic UDI-DI
- UDI (consisting of UDI-DI and UDI-PI)
- Packaging UDI (Note: This is not an official term used in the EU MDR and IVDR, but we’re using it to help explain the concept. The Packing UDI is part of the UDI itself.)
1. Basic UDI-DI
The Basic UDI-DI identifies the device group that a particular device fits into. A device group is a group of products that all share the same intended purpose, risk class, essential design, and manufacturing characteristics. A device group is generally classified by medical device manufacturers as a “Product Family” or “Product Category,” depending on the internal nomenclature used within the company. The Basic UDI-DI functions as a parent or higher-level descriptor of a device.
NOTE: There can only be one Basic UDI-DI per UDI-DI.
The Basic UDI-DI is not printed on the product itself or on the packaging of a product, but rather it must be included in the following documents and applications:
- Certificates (Including Certificate of Free Sale)
- EU Declarations of Conformity
- Techical Documentation
- Summary of Safety and Clinical Performance
2. UDI (UDI-DI and UDI-PI)
The second component is the UDI itself, which consists of two parts:
Device Identifier (DI)
Production Identifier (PI)
The UDI-DI (Device Identifier DI, also referred to as “static”) identifies specific, detailed information about a particular device. If any of the below details should change, the device will need a new UDI-DI.
- Name or trade name of the device
- Device version or model
- If labelled as a single use device
- Packaged as sterile
- Maximum number of uses
- Need for sterilization before use
- Quantity of devices provided in a package
- Critical warnings or contra-indication
- CMR/endocrine disruptors
NOTE: There can be several UDI-DIs for one Basic UDI-DI.
Meanwhile, the UDI-PI (Production Identifier PI, also referred to as "dynamic") contains manufacturing information (including serial number, lot/batch number, software identification, and manufacturing or expiry date or both types of dates.)
To better illustrate this concept of Basic UDI-DI and UDI (UDI-DI and UDI-PI), let’s use a syringe as an example. The Basic UDI-DI would identify the category of a syringe, for example, "Enteral (Oral) Syringe."
A 5ml Enteral (Oral) Syringe – Sterile (Color: Purple) would get a unique UDI-DI and a 10m Enteral (Oral) Syringe – Sterile (Color: Orange) would get a unique UDI-DI. Both products would be associated to the same Basic UDI-DI. In this case, the "Enteral (Oral) Syringe," which defines the category.
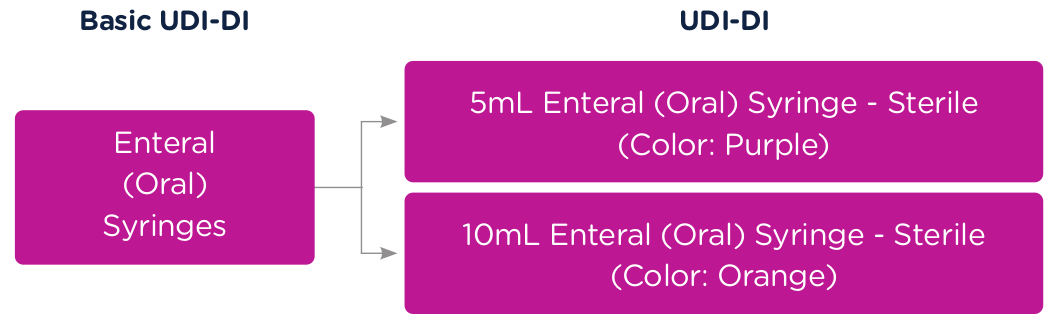
Each time that 5ml Enteral (Oral) Syringe – Sterile (Color: Purple) is manufactured at the same revision, it will get a new UDI-PI per lot. See the graphic below.

Each product is identical and therefore has the same UDI-DI. However, the UDI-PI changes to reflect the manufacturing date, lot number, expiry date, and serial number, as applicable.
The UDI will contain all device-specific information and have the same functions as the comparable database (GUDID) of the United States FDA. The main difference (in EUDAMED) is that the UDI data is divided into components of Basic UDI-DI, UDI, and Packaging UDI.
3. Packaging UDI
The third component of UDI is the Packaging UDI. (Note: This is not an official term used in the EU MDR and IVDR, but we’re using it to help explain the concept.)
Each level of packaging, except shipping containers, must receive its own unique UDI. Packaging UDI refers to the unique UDI assigned to higher levels of packaging instead of the device itself.
In the event of significant space constraints on the unit of use packaging, the UDI Carrier may be placed on the next higher packaging level.
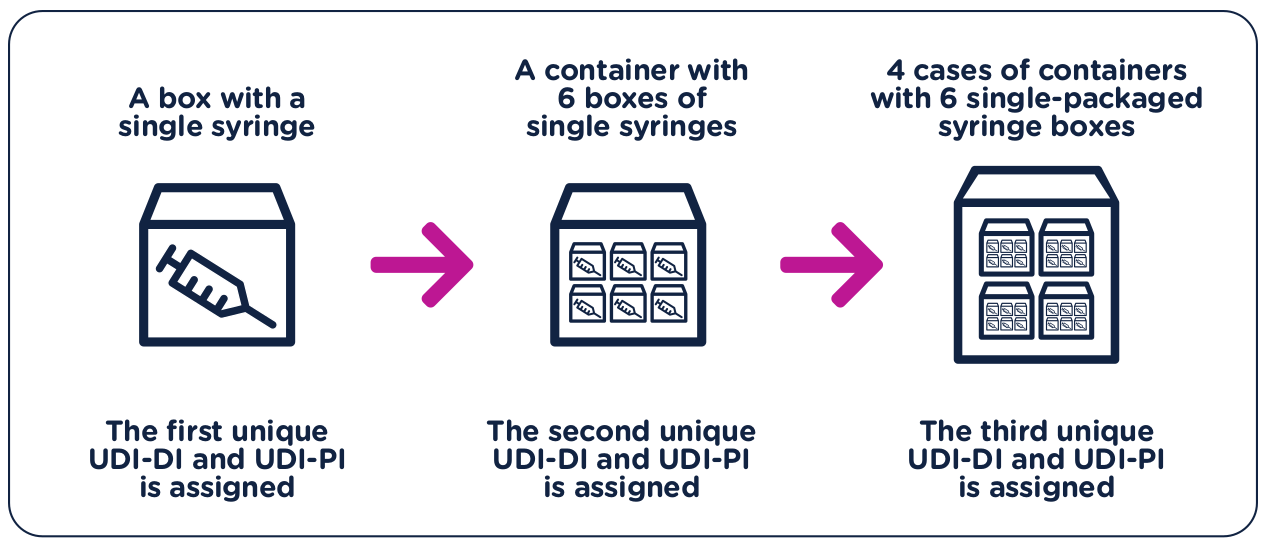
Returning to our earlier example of syringes, if a manufacturer first packages a single sellable syringe into an individual box, this package would receive its own UDI-DI and UDI-PI.
If then the manufacturer packages those individual boxes into containers of six (6), those containers would receive their own UDI-DI and UDI-PI.
And finally, if the manufacturer packages those six (6) containers into cases of four (4), those cases would receive their own UDI-DI and UDI-PI.
Each of those levels of packaging must be assigned its own UDI-DI and UDI-PI. The initial syringe did not change, but the way it is packaged did, therefore, requiring its own UDI-DI and UDI-PI.
Element 2: Placing UDI on the device and/or packaging
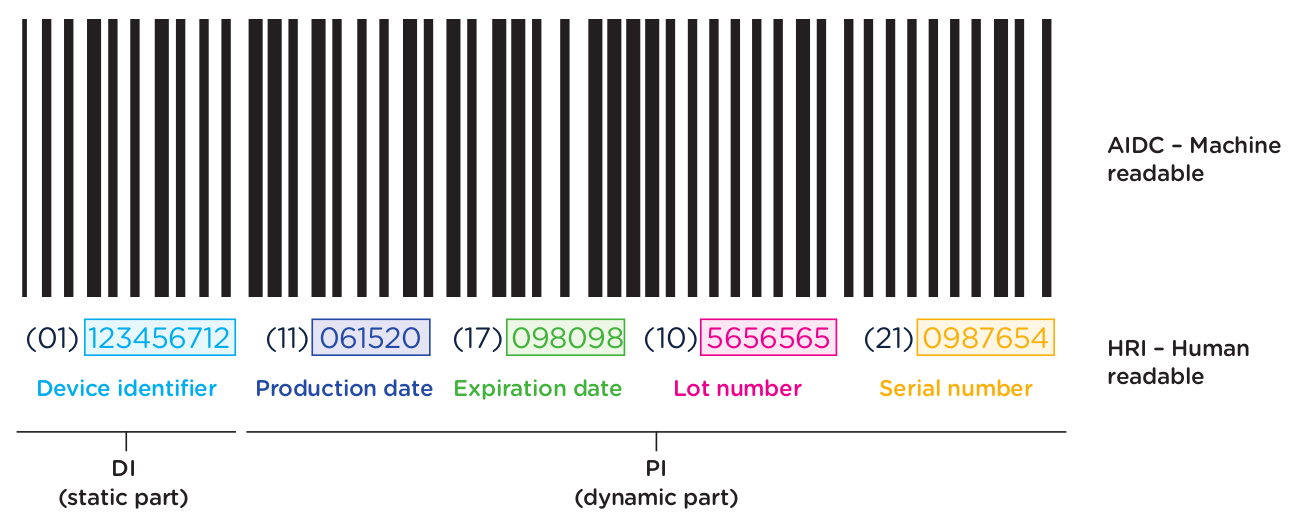
The second element to the UDI system is the placing of the UDI on the device or on its packaging through what is referred to as a “UDI Carrier.” The UDI Carrier is the part of the label that contains the UDI information that is applied directly to the device or included on the device packaging. The UDI Carrier should have both a machine-readable portion (AIDC) and a human-readable portion (HRI). (Specific details about each element of the UDI will be covered in Chapter 2.)
- Machine-readable form – AIDC – (Automatic Identification and Data Capture) is a barcode or other machine-readable technology that can be accessed automatically by scanning the UDI information.
- Human-readable form – HRI – (Human Readable Interpretation) is the numeric or alphanumeric code, which can be manually entered into the system for access to the UDI information.
If there are space constraints limiting the use of both the AIDC and HRI on the label, then only the AIDC is required to appear. However, on devices that are intended to be used in home-health care or other non-medical facility settings, the HRI would be required to appear.
Single-use devices may contain the UDI Carrier on its lowest level of packaging rather than on the device itself.
Reusable devices must include the UDI Carrier on the device itself, unless any type of direct marking would interfere with the safety or performance of the device, or if it is not technologically feasible to directly mark the device. If so, this should be properly documented in your design history file.
Most importantly, the UDI Carrier must be readable for the intended lifecycle of the device.
Below is an example of a GS1 AIDC and HRI barcode label.
Element 3: Storage of UDI information by Economic Operators
Storage of UDI information by "Economic Operators" is the third element of the UDI system. 2017/745 Articles 2(35), 22(1), and 22(3) define an economic operator as:
- A manufacturer
- An authorized representative
- A distributor
- An importer
- An investigator for clinical investigations
- A person who sterilizes systems or procedure packs
Class III, implantable device:
According to EU MDR 2017/745 Annex II, the manufacturer shall keep an updated list of all UDIs that it has assigned. Economic operators and all health institutions are required to store, preferably by electronic means, the UDI of all the devices for which they have supplied or with which they have been supplied.
For Devices Other than Class III:
Member States are encouraged, and in some cases require, health institutions to store, preferably by electronic means, the UDI of the devices with which they have been supplied. The UDI must also be included in any field safety notice for reporting serious incidents and field safety corrective actions.
The EU MDR and EU IVDR also give the European Commission authority to make additional requirements regarding the submission or maintenance of UDI information. In making those decisions, the European Commission must consider six (6) areas:
- Confidentiality and data protection
- Risk-based approach
- Cost-effectiveness of the additional measures
- The need to avoid duplications in the UDI system
- The needs of the healthcare systems of the member states
- Harmonization with other medical device identification systems
To continue reading this eBook including information about the EUDAMED database, UDI format requirements and issuing entities, implementation timelines, and key differences between the EU and US UDI systems, please register to download the full version


GET IN TOUCH






.avif)
