
What is BUDI?
By now, you should be familiar with the terminology surrounding UDI - The Unique Device Identification System. The United States FDA, the European Commission, and other regulatory bodies around the world have developed UDI regulations for medical devices and in vitro diagnostic devices that involve both labeling and database registration requirements. In the EU, UDI regulations were introduced under Regulations (EU) MDR 2017/745 and (EU) IVDR 2017/746. There is UDI, UDI-DI, UDI-PI - so then what is a BUDI-DI?
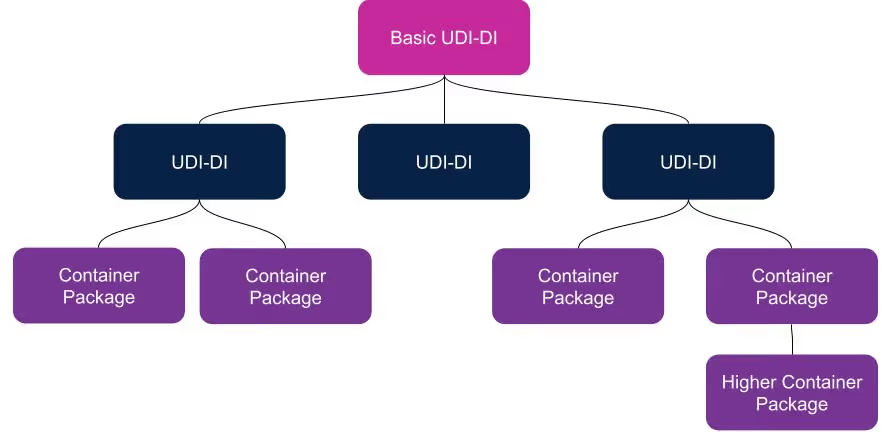
BUDI is an abbreviation for “Basic UDI” and is commonly pronounced “Buddy.” A BUDI-DI is unique to the EU and allows devices with multiple UDI-DI’s to be grouped together. It is necessary whether you have one device group (sometimes referred to as device ‘family’) or have many different device configurations such as systems, procedure packs, or kits. The general rule is there can only be one BUDI-DI to many UDI-DI’s and never multiple BUDI-DI’s to just one UDI-DI. The only time a BUDI-DI is not required is for a custom-made device, which generally doesn’t fall into the UDI requirements of the MDR/IVDR anyway.
A BUDI-DI allows manufacturers to connect and identify device groups with the same intended purpose, risk class, essential design, and manufacturing characteristics. It is an identification number that is only used for administrative purposes. It is required in the EUDAMED database and is referenced in relevant documents such as certificates, declarations of conformity, and technical documentation. If the device requires Notified Body review, then the BUDI-DI should also be listed on the CE Certification and the Certificate of Free Sale.
A BUDI-DI is the key that unlocks the EUDAMED and provides access to all of the product information.
- It’s the primary identifier of the device group/family
- It’s the main record key in the EUDAMED
- It’s the main product identifier in the regulatory documentation
- It’s independent of packaging and labeling
UDI issuing agencies
The manufacturer is legally responsible for utilizing a human-readable BUDI-DI assigned by an approved UDI issuing agency, such as HIBCC or GS1. The format of the BUDI-DI will vary slightly depending upon which issuing agency you work with. Currently, the only approved issuing agencies in Europe are GS1, HIBBC, ICCBBA, and IFA.
Per the MDCG 2019-1 guideline, each agency must:
- Create a code format that is close to the existing UDI-DI format
- Use no more than 25 total characters
- Assign a check/digit character that was determined by an algorithm
A BUDI-DI cannot be changed. A product UDI-DI created because of a new product variation or changes to the UDI-DI data elements can report into an existing BUDI-DI.
The EU provides an EU UDI Helpdesk to assist with navigating UDI requirements and answering questions device manufacturers may have.
Note that EUDAMED registrations, including BUDI-DI numbers, are currently recommended but not required. Use of the EUDAMED databases will not become required until all six databases are live, which is expected to be in Q2 of 2023, with a 24-month transition period.
Read our Quick reference guide - global medical device UDI requirements and timelines for additional information on general UDI requirements.

GET IN TOUCH






.avif)
