
This article is an excerpt from The ultimate guide to the China NMPA UDI system and database ebook.
Table of Contents
- Overview
- UDI basics and benefits
- UDI format requirements and issuing entities
- UDI database and submission requirements
- Implementation of UDI and the UDI database in China
The current Chinese medical device regulatory regime kicked-off in 2014 with the Regulation on Supervision and Administration of Medical Devices. This core set of registration requirements, modeled after the United States and European Union systems, established a set of device classifications (class I, II, and III) based on risk and procedures for obtaining market clearance for each type of device.
Medical devices in China are regulated by the National Medical Products Administration (NMPA). Class I devices, such as clinical laboratory equipment or non-invasive skin dressings, require only notification to the NMPA for marketing authorization, and that authorization does not expire. Class II and III devices such as implantable devices or devices with a measuring function require full registration and a formal review before market clearance can be obtained.
These initial regulations have been expanded since their introduction, adding accelerated pathways to market for certain products in certain regions, easing acceptance of clinical data from overseas, and more specific roles and responsibilities for local agents of international manufacturers. In addition, in 2019, the regulations added a provision that medical devices carry a unique device identification (UDI). China’s UDI requirements are similar to those in the US and European Union. They establish specific device ID and labeling requirements, as well as a central, state-administered database of devices.
This eBook walks through the basics of medical device UDIs, the specifics of China’s implementation, and how MedTech companies who market their devices in China can prepare for the full rollout of these regulations in the coming years.
A UDI is a unique alphanumeric code that is designed to identify medical devices sold in a particular country/region from manufacturing, through distribution, to use by a patient. Like other aspects of the medical device regulatory regime, the UDI system in China follows the approach taken by the United States FDA and European Commission, and is based on the guidance from the International Medical Device Regulators Forum (IMDRF). Generally, UDI systems are designed to improve patient safety and optimize care by:
- Increasing the traceability of medical devices, including field safety corrective actions
- Providing an unambiguous identification method for medical devices throughout distribution and use
- Making adverse event reports more accessible
- Reducing medical errors by providing detailed information related to the device
- Simplifying medical device documentation and making it more consistent
There are three components to the UDI system in China:
- UDI code: The actual UDI code can be assigned by one of three (3) issuing agencies and contains information about the product, it’s expiration date, and the manufacturing batch/lot it’s associated with.
- UDI labeling: Put simply, medical devices must carry the UDI code on them. The regulations stipulate how devices and their packaging must be labeled for compliance.
- UDI database: In addition to labeling, all device UDIs must be submitted to a central database that is administered by the NMPA.
The following sections explore each of these components in more detail.
The UDI code
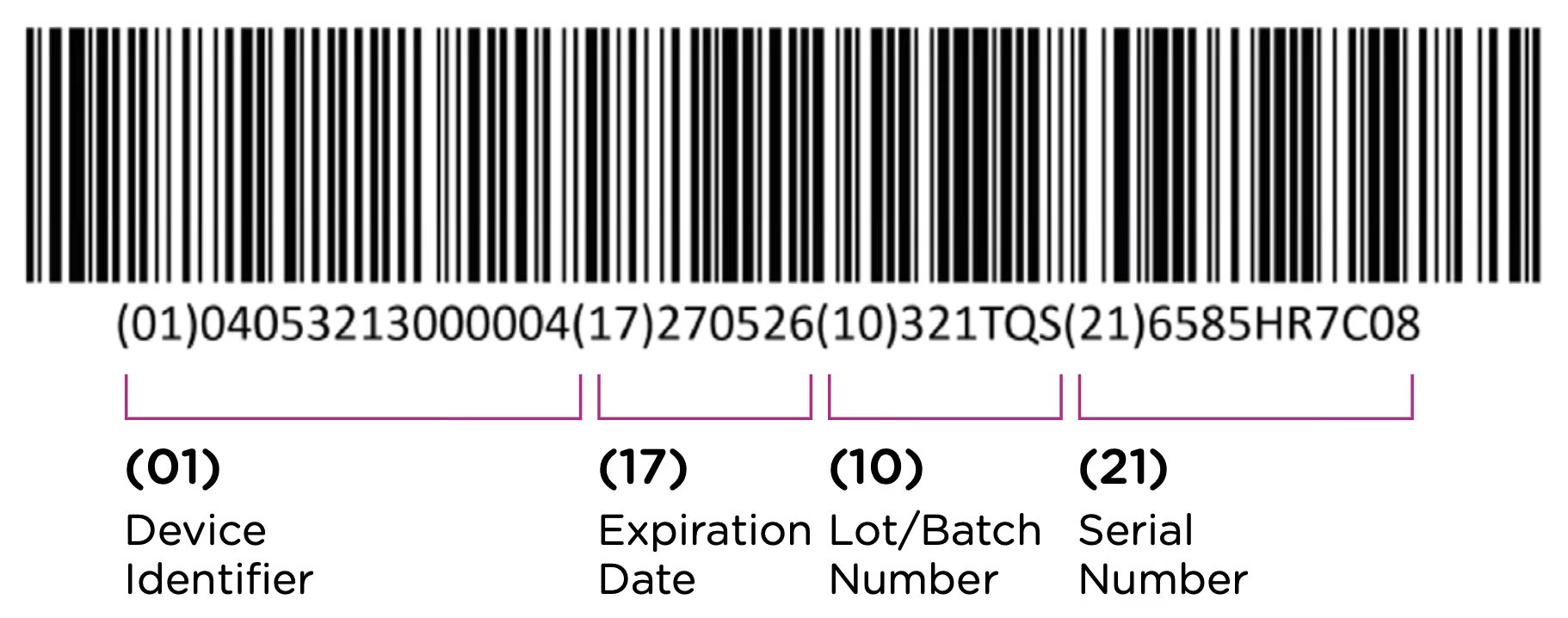
The first element of the UDI system is the code itself. The UDI code is the alphanumeric identifier that is associated with a specific medical device. UDI codes have two (2) elements to them, the UDI device identifier (UDI-DI) or static portion, and the UDI production identifier (UDI-PI) or dynamic portion. You can see the two components in the UDI diagram below:

The UDI-DI contains information about the issuing entity—the organization that is authorized to assign UDI codes. In China, this can be one of three entities: GS1, an international barcode and electronic data interchange standards organization, and two domestic organizations: the Zhongguancun Industry & Information Research Institute (ZIIOT), and AliHealth. Additional details about the issuing agencies are covered in Chapter 2. In addition, the UDI-DI contains information about the manufacturer and the specific model or version of the device.
The UDI-PI contains information about the manufacturing and production of the device. This typically includes information about the lot or batch number in which the device was manufactured, the manufacturing date and expiration date for the device (if applicable), and the specific serial number for the device. Here you can see all of the components marked up using the same UDI example:
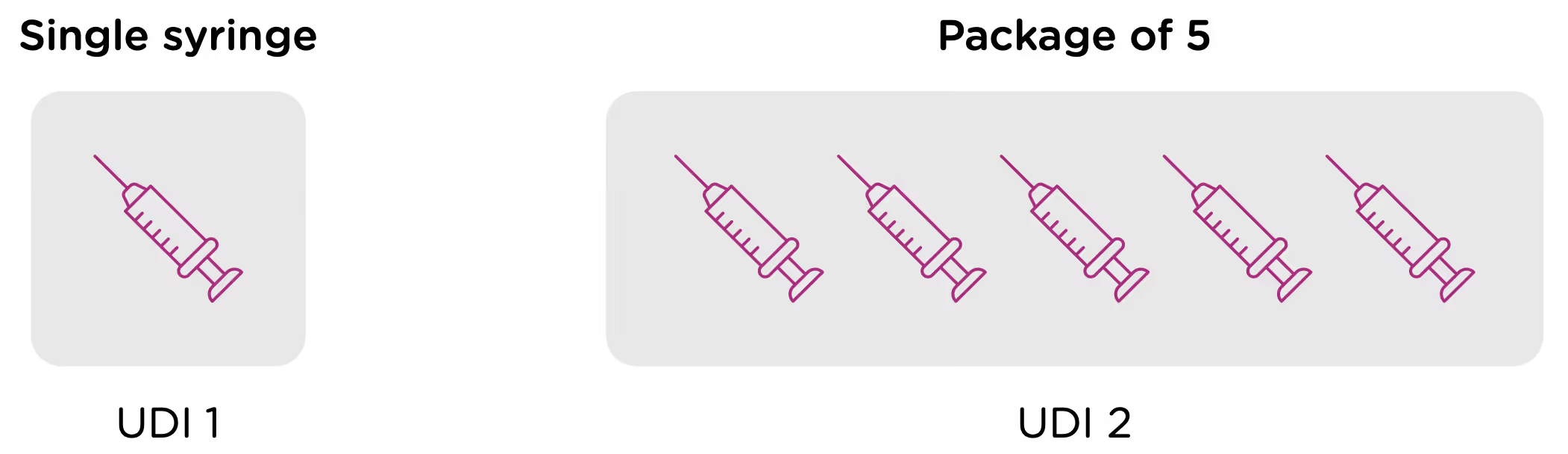
Note that each packaging permutation and level for a given device will need to be assigned its own UDI. So for example, let’s say that a company manufactures 5ml enteral (oral) syringes in two packaging options: 1 – packaged individually and 2 – packaged in a box of 5. Each packaging option would need its own UDI, despite the fact that the underlying product is the same.
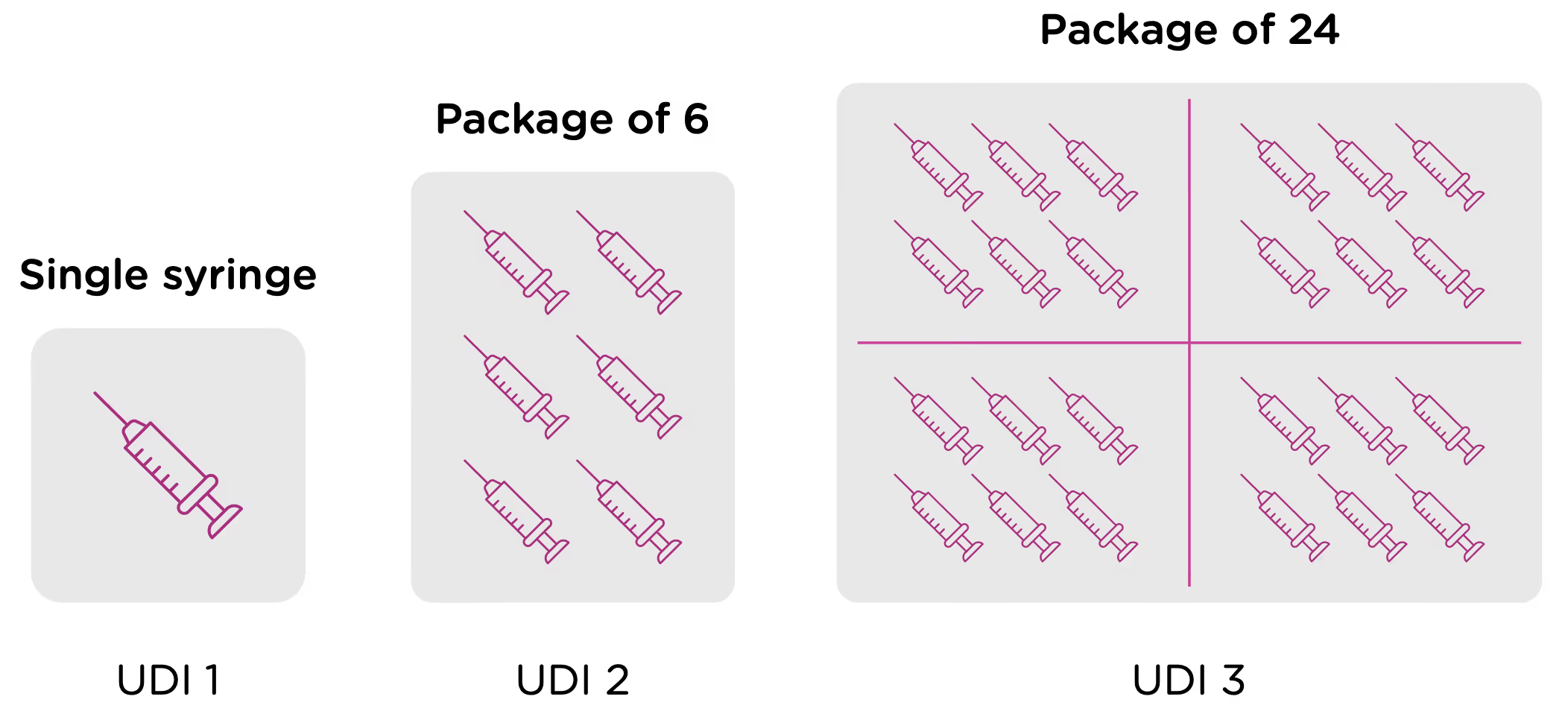
Now looking at packaging levels, let’s assume that the manufacturer packages the single syringe offering into boxes of 6, and again into larger containers of 24. Each of those packaging options needs its own UDI as well.
Labeling
In addition to obtaining UDI code for each device as outlined in the previous section, medical device manufacturers are required to ensure that devices are appropriately labeled with the assigned UDI. This label is called the UDI Carrier. The UDI is represented in two forms on the UDI Carrier: a machine-readable form and a human-readable form.
The machine-readable form or automatic identification data capture (AIDC) is a barcode or some other technology that can be used to automatically capture UDI information. The NMPA regulations support 3 types of machine-readable formats: 1-dimensional barcode, 2-dimensional barcode, and radio-frequency identification (RFID).

The regulations note that “use of advanced automatic identification and data collection technologies is encouraged”—prompting manufacturers to use more modern 2D and RFID machine-readable carriers where possible. Note, however, that if a device uses RFID, the UDI Carrier must also include the UDI in barcode format.
The human-readable form or human-readable interpretation (HRI) is the numeric or alphanumeric code for the UDI that can be read and manually entered into systems.

The UDI Carrier should be included on the device and on all levels of packaging. The UDI Carrier must be clear and readable during the operation and use of devices. If there isn’t room on the device for both the human and machine-readable forms of the UDI, then manufacturers should prioritize the machine-readable form.
UDI database
The third component of the NMPA UDI system is the UDI database. This is a centralized database of UDI and product information, administered by the NMPA. Manufacturers are required to submit UDI information into the database within 60 days after a product is approved (for sale in China) and before it is commercialized. The database contains a more detailed product record than what is included in the UDI itself, and it is the responsibility of the manufacturer (and/or their in-country representative) to submit the information correctly, and ensure that it’s kept up to date.
Chapter 3 of this eBook goes into detail about the specific fields and data requirements for UDI database submissions.
To continue reading this eBook including information about UDI format requirements and issuing entities, implementation timelines, and affected device types, please register to download the full version.


GET IN TOUCH






.avif)
