
This article is an excerpt from The beginner's guide to the FDA PMA submission process ebook.
Table of Contents
- Introduction
- PMA basics
- FDA interactions
- Contents of a traditional PMA submission
- PMA supplements and amendments
- PMA Quality Management System (QMS)
- Review process and timeline
If your organization is planning to market a new medical device in the United States, you first need to determine which regulatory class the device falls under. The vast majority of medical devices regulated by the FDA are either Class I or Class II medical devices, requiring a 510(k) premarket notification or a simple registration if exempt from 510(k) requirements. However, if your device sustains or supports life, is implanted, or presents a “potential unreasonable risk of illness or injury,” your device is likely a Class III device which will require Premarket Approval (PMA) from the FDA before it can be marketed in the United States. Novel devices, for which there are no existing substantially equivalent devices, are automatically classified as Class III as well. Novel devices with a lower risk profile, however, may qualify for the De Novo process instead of the PMA. Just 10% of devices regulated by the FDA are Class III devices.
This ebook provides an overview of the PMA process and its requirements, but it is not designed to be the only resource used in compiling a PMA submission. The FDA provides significant documentation on this process, starting with the regulation governing premarket approval that is located in Title 21 Code of Federal Regulations (CFR) Part 814.
FDA: Background and device oversight
Before we explain what a PMA is, let’s first talk generally about the Food and Drug Administration (FDA) and device oversight. The FDA is the U.S. governmental agency responsible for overseeing medical devices, drugs, food, and tobacco products. When it comes to medical devices, the FDA’s mission is to “protect the public health by ensuring the safety, efficacy, and security of...medical devices.” At the same time, the FDA also has an interest in “advancing public health by helping to speed innovations.” In other words, the FDA’s goal is to make sure devices are safe and effective for public use, while also ensuring that devices have a quick and efficient path to market.
In order to achieve this balance of safety and efficiency, the FDA has three different levels of oversight depending on the risk level of the device: (1) exempt from premarket notification, (2) Premarket Notification, also known as 510(k), and (3) Premarket Approval (PMA).
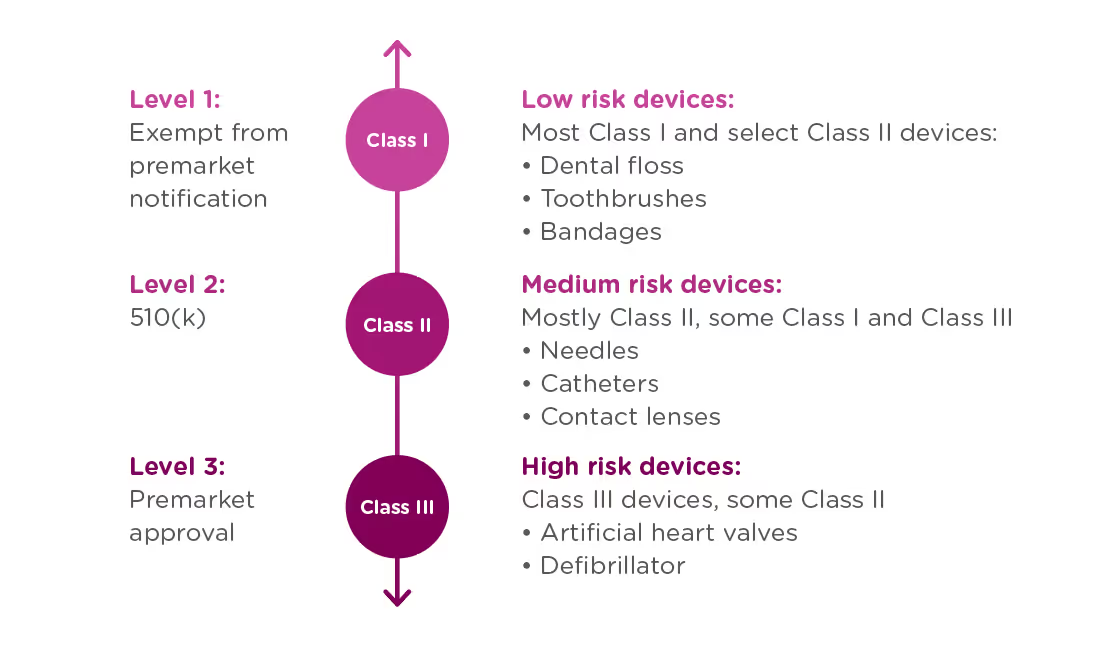
When is a PMA required?
The PMA process is the most stringent regulatory process for medical device approval under the FDA and applies to almost all Class III devices. To determine whether your device requires a PMA, you must first Classify your device by searching the Product Classification Database. The database will provide you with similar devices; their name, classification, and link to the Code of Federal Regulations (CFR) if applicable.
- If a substantial equivalent is found in the Product Classification Database with a submission type of 510(k), you should submit a 510(k), not a PMA.
- If the product classification database identifies your device as Class III and/or requiring a PMA - you should submit a PMA.
- If your device involves a new concept and does not have a classification regulation in the CFR, the database will list only the device type name and product code. In this case, the three-letter product code can be used to search the PMA database and the 510(k).
- If your device cannot be found in the product classification database because it is a new type of device and should be classified as a Class III device because of the level of risk it presents*.
Class III devices support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential and unreasonable risk of illness or injury.
Note that if your device is a new concept without a substantial equivalent, but does not present the level of risk of a class III device, it may be eligible for the De Novo process as a class I or class II device.
PMA vs 510(k)
Not only are PMA and 510(k) processes applicable to different types of devices, they have different purposes.
510(k): A 510(k) is intended to demonstrate that the device for which approval is being sought is as safe and effective as a currently marketed device that does not require a PMA.
PMA: A PMA is intended to prove that a new device is safe and effective for the end user. A PMA is much more detailed and in-depth than a 510(k). Device manufacturers are typically required to present human clinical trial data, in addition to laboratory testing data.
The difference in complexity between a PMA and 510(k) also affects the time needed to process the submissions. The FDA typically accepts or rejects a 510(k) submission within 30-90 days, at which point the device is posted to the FDA’s 510(k) database. A PMA submission can take up to 180 days to be processed, at which point the FDA can approve or deny the application. The FDA may also issue an “approvable” or “not approvable” letter, which the applicant can choose to respond to, thereby adding time to the submission process.
PMA application methods
There are a number of types of PMA application methods. While most devices which require a PMA will follow the traditional process, be sure to verify that you are using the correct application process to maximize your chances for success and avoid unnecessary delays:
Traditional PMA
The most common method for attaining FDA clearance for Class III devices, the traditional PMA is the appropriate option for most devices that have completed clinical testing.
Modular PMA
The modular PMA is the appropriate application method for devices that have not yet completed clinical testing. Applicants complete individual “modules,” with final confirmation granted once all sections are completed. For additional information on specific requirements of a modular PMA, read the FDA’s Premarket Approval Application Modular Review.
Product Development Protocol
Use the Product Development Protocol (PDP) with medical devices that are based on well-established technology. The PDP process for gaining market approval merges the clinical evaluation and development of information, and involves an agreement between the manufacturer and the FDA. The process provides the advantage of early predictability for the manufacturer and allows early interaction that can identifyFDA concerns as soon as possible in the development process. Because the PDP identifies the agreed upon design and development details, a completed PDP is considered to have an approved PMA. For additional information, read more about the FDA’s PMA Application Methods.
Humanitarian Device Exemption
A Humanitarian Use Device (HUD) is specifically defined as a device intended to benefit patients that are affected by a disease or condition that affects less than 8,000 individuals in the U.S. per year. TheHumanitarian Device Exemption (HDE) approval process is designed to encourage clinical activity around rare conditions, and does have certain restrictions, including:
- After receiving HDE approval, a HUD is eligible to be sold for profit only if the device is intended to address a disease or condition that occurs primarily in pediatric patients, or occurs in pediatric patients in small numbers.
- If an HDE is approved to be sold for profit, the FDA will determine an annual distribution number(ADN). Any devices sold beyond the ADN limit are required to be sold for no profit.
For more information see the FDA’s explanation of the Humanitarian Device Exemption.
CBER Submissions
There are two centers within the FDA responsible for evaluating medical devices. While the majority of devices will go through the Center for Devices and Radiological Health (CDRH), some will be managed by The Center for Biologics Evaluation and Research (CBER). CBER regulates medical devices related to blood and cellular products, including blood collection and processing procedures as well as cellular therapies. This ebook focuses on submissions made through the CDRH, but you can view CBER Regulatory Submissions – Electronic and Paper for more information on the CBER process.
To continue reading this eBook, including a walk through of the different types of required and optional FDA meetings and communications, a detailed list of the contents of a traditional PMA submission, and an overview of quality management system requirements, please register to download the full version.


GET IN TOUCH






.avif)
