
While it may seem simple, medical device classification can be a challenging task for many medical device and IVD manufacturers. Device classes for specific regions and countries have a number of small variations, and each of those variations can impact the process by which a device obtains market clearance. Getting it wrong can lead to delays in getting to market. This article explores the classification systems for three major markets, and their associated regulations.
An important component of achieving regulatory approval is the appropriate classification of a medical device or in vitro diagnostic device, according to the specific regulations within a country or region. Product classifications are related to the intended use of the product and the perceived risk that it poses to a patient using the device. While this general approach is pretty standard across all regions, there are many small variations in different country classification systems that can impact how a device is regulated. It would be much easier if there was one global classification system that everyone followed.
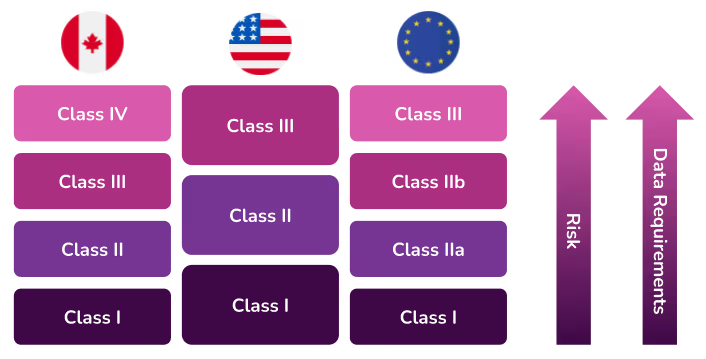
However, since there are different guidelines to classifying a medical device (per country), we’ll dig into the most popular classification systems—Canada, the European Union and the United States. These three are globally perceived to have strong, thorough, and trusted quality and regulatory systems. Their approaches are often mirrored or used as proxies for market clearance in other countries.
Canada
The Canadian Medical Devices Regulations include guidelines that classify devices into four risk classes. If a medical device can be classified into more than one class, the class representing the higher risk always applies.
- Class I devices do not require a medical device licence to be sold in Canada, but manufacturers, distributors and importers of these devices are required to obtain an establishment licence.
- Class II requires a medical device licence
- Class III requires a medical device licence
- Class IV requires a medical device licence
Some examples of different classes of devices in Canada include:
European Union
One of the main changes introduced with the new MDR/IVDR regulations are new classification rules for medical devices and in vitro diagnostic devices. If you have gone through the process of getting your medical device in the European market before, you might find it more difficult with the new EU MDR rules. For example, a new medical device you want to bring to market might now fall into a higher classification than it would have previously (under the MDD), and therefore require more testing, updates to documents, quality approvals, etc.
The new EU MDR brings the classification of medical devices in Europe more in line with international regulations, specifically the United States. These updated rules are listed in MDR 2017/745 for devices and IVDR 2017/746 for in vitro diagnostic products. As with Canada, if a medical device can be classified into more than one class, the class representing the higher risk always applies.
The EU has recently released a new guidance document MCDG 2021-24 to assist device manufacturers with device classification questions.
Class I – this classification is for the lowest risk device. Most medical devices in this category do not require a conformity assessment from a Notified Body so instead, they can be self-assessed. However, manufacturers must still complete a Technical File as part of the approval process.According to MDCG 2019-15, there are three subclasses under Class I. Unlike most Class I devices, these will require the involvement of a Notified Body.
- Class Im: a product with a measuring function
- Class Is: a product that is sterile
- Class Ir: a product that is a reusable, surgical instrument
Class IIa – this classification is for a medium risk device. A conformity assessment by a Notified Body is required for this classification.
Class IIb – this classification is for medium-to-high risk devices. A conformity assessment by a Notified Body is required for this classification.
Class III – this classification is for the highest risk devices. A conformity assessment by a Notified Body is required for this classification.
Examples of different classes of devices in the European Union include:
In Vitro Diagnostic Devices:
Class A – this classification is for the lowest risk in vitro diagnostic devices. Most IVD devices in this category do not require a conformity assessment. Instead, they can be self-assessed.
Class B – this classification is for medium risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Class C – this classification is for medium-to-high risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Class D – this classification is for the highest risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Examples of the different in vitro diagnostic device classes in the European Union include:
United States
In the United States, the Food and Drug Administration (FDA) is responsible for overseeing the safety of medical devices. The FDA has established classifications for approximately 1,700 different types of devices and grouped them into 16 medical specialties referred to as panels. All three classes of devices are subject to General Controls which are the baseline requirements of the Food, Drug and Cosmetic (FD&C) Act.
- Class I – General Controls (with or without exemptions)
- Class II – General Controls and Special Controls (with or without exemptions)
- Class III – General Controls, Special Controls and Premarket Approval
You are permitted to classify your own medical device based upon the FDA guidance documents and set regulations. However, if you wish for the FDA to assist with establishing your classification you can submit a 513(g) Request for Information. Note that there is a user fee associated with a 513(g) Request.
The device class determines which type of premarketing submission/application is required for market clearance.
In some instances, you do have the opportunity to reclassify your product after it’s been released to the market. The regulatory class of a device type, as defined by the Federal Food, Drug and Cosmetic Act (FD&C Act), may be changed through petition to the FDA. This process is only applied to a device type though, not to an individual device.
Examples of medical device classification in the US include:
Getting classification correct
Medical device classification is simple in that each country and region generally follows the same classification approach, and complex in that minor differences can change how a device is classified across markets. Understanding how a device is classified is one of the critical first steps regulatory affairs teams need to take when entering a new market, as medical device class often determines the pathway to market.
For example, in the EU classification can mean the difference between self-certification and a required conformity assessment from a Notified Body. In the US, classification can mean the difference between a 510(k) or PMA process for market clearance. Getting classification correct can ensure a smoother and faster route to market.
To learn more about market clearance processes for medical devices in the US, check out the Beginner’s Guide to the 510(k).


GET IN TOUCH






.avif)
