
Software for medical device regulatory teams
Many regulatory affairs (RA) teams within medical device organizations are still managing their activities through spreadsheets, in-house custom-built software, or systems designed for other purposes. We believe that regulatory teams deserve purpose-built software that allows them to ensure compliance across products and markets, and provides them with the opportunity to contribute directly to revenue-driving activities.
Regulatory Information Management (RIM) solutions provide the centralized regulatory functionality needed by today’s RA teams. RIM solutions such as Rimsys are product-centric, allowing regulatory professionals to track all product-specific information and then create market submissions, link standards and essential principles, manage registrations by product by market, and control all regulatory approvals and projects. However, not all RIM solutions are created equal. If you have complex products, devices that include software, or other requirements that not all medical device companies will have, be sure to carefully evaluate potential systems for their ability to address those needs.
Justifying the need for RIM
Any software selection project begins with analyzing the need for the new software, creating a justification for the project, and obtaining the approval and budget to move forward. RIM solutions will allow your RA team to find information more quickly and operate more efficiently, which means that justification for a new RIM system typically comes from four areas:
- Cost savings – RA teams can operate more efficiently, reducing the need for outside consultants or contractors and enabling new RA team members to onboard more quickly. Better information also allows RA teams to better forecast projects in order to optimize the internal team size.
- Reduced regulatory risk – A centralized RIM system reduces multiple risks, including missing an expiration date or supplying incorrect information to a regulatory body. Even a small misstep can cause an audit finding, removal of a product from a market, or a delay in entering a new market.
- Improved competitive advantage – RIM systems significantly reduce the time that RA teams spend finding data and reacting to last-minute “emergency” requests. This advantage allows the team to drive competitive advantages and greater revenue growth through participation in market planning, product roll-out decisions, and other strategic planning. One Rimsys customer was able to improve the time for a product release by 88%. By increasing speed to market, medtech companies can recognize revenue sooner and capture more market share more quickly.
- Data harmonization – Medtech companies dealing with duplicated data and systems due to mergers or acquisitions can point to a centralized RIM system as a way to harmonize important regulatory data to ensure compliance and optimize go-to-market activities.
You will likely need to develop a comprehensive business case to support any RIM investment. This will include detailing the limitations of current approaches (including spreadsheets and costs to maintain in-house systems), quantifying the expected benefits, and explaining the evaluation process to arrive at your preferred vendor. Building this as you work through the early stages of RIM selection will prevent delays as you move into the purchase process.
Should I use external consultants to help with RIM selection?
RIM selection projects are sometimes managed by in-house teams and sometimes managed by 3rd-party consultants. How do you decide which is right for your selection project?
Does your team have experience with large system selection projects?
If your organization has a large IT team and/or digital transformation team, they likely have the responsibility of overseeing the selection of any new software systems. Be sure to understand their capabilities – have members of the team managed large system selection projects before, such as ERP or PLM selections? The regulatory team and others within the organization can provide subject matter expertise, but you will be relying on the technical team to oversee the project, define requirements, help set a budget, and more.
If the right level of expertise does not exist within your organization, an outside consultant with medical device regulatory experience and with business system selection projects should be considered. This type of consultant can be extremely helpful during the system implementation and adoption phase of the project as well.
Does our team have the bandwidth to manage a RIM selection project?
Even if your organization has an internal team with the expertise to manage a RIM selection project, they may not have the time to do so within the desired project timeframe. In this case, an external consultant can augment your existing team to get the project completed as required.
Do we need a new perspective?
Selecting a RIM solution is as much about digital transformation and process optimization as it is about ensuring you find a system with the right features. Do you have a vision of where you want the RA team to be? Have you looked at the characteristics of top-performing RA teams? If you think you might need a new perspective and an outside voice, an outside consultant may be the right choice for your project.
RIM selection project steps
Once you have determined the need for a RIM system, a selection project should include the following major steps:
Build the selection team
Put together a core selection team that consists of:
- A project leader that is typically at a manager level or above within the organization and comes from the regulatory or IT teams.
- An executive sponsor who may not participate in all aspects of the project, but who will ensure that resources are made available to the team as needed and that the project is kept on track and is aligned with overall company goals.
- IT team member(s) who will provide feedback on technical system requirements, including security and data privacy, and will support customization and integration discussions.
- Subject matter experts representing each department and team who will be using or interacting with the software.
You will add team members once you begin to implement the system, but selection teams typically consist of fewer than 10 members.
Determine requirements and selection criteria
One of the primary responsibilities of the selection team is to define the requirements for the new system, and the criteria on which systems will be judged. Requirements usually fall into multiple categories, including:
- Business requirements – These are broad requirements based on business needs. They will typically answer the question “Why do we need a RIM system?” For example, reducing the administrative burden on the regulatory team with functionality that allows the team to track registration expiration dates, create submission dossiers, and quickly report on registration status by product and market. Include project timeline and budgetary constraints here as well.
- Functional or user requirements – Functional requirements are a more detailed list of specific functions that users will need to perform in the system. For example, the ability to link standards to essential principles, manage multiple approvals for submission documents, or track UDI product data. DO include requirements that are essential for users to complete their work within your defined quality procedures. DO NOT include requirements that are unnecessarily specific (ex: the product description must contain at least 50 characters) or are so common that all systems will meet the requirement (ex: there must be a product number and description in the product record).
- System and technical requirements – Your IT team may already have a list of the overall technical requirements that all software must meet within your organization. These will likely include data security requirements and features that support system validation. Include any specific requirements regarding system availability, upgrade management, and technical support guarantees. See SaaS 101 for medtech regulatory professionals for a list of questions that you should ask a SaaS solution provider. Also include here any procurement requirements that your organization has, such as insurance requirements.
- Vendor resources and vision – You want to work with a vendor that shares your vision, not only for this system implementation but also for future growth. Evaluate each vendor’s product roadmap and plans for innovation against your organization’s digital transformation plans for the next 2-5 years. Does the vendor share your vision? Do they have the resources to support your organization and that future vision?
Establish project goals and timelines
Establish project goals and an overall project timeline. Is there a hard deadline by which the system needs to be live? What are the goals and metrics with which the success of the project will be measured? Be sure to get written agreement from the project team and executive team on the goals, timeline, and how the information will be reported.
Research RIM vendors (build your initial list)
If you are working with an experienced regulatory consultant, they may be able to get you to your short list without this step. However, if you are unsure of which systems may meet your needs, begin by:
- Researching Regulatory Information Management systems online.
- Talking to other regulatory professionals for suggestions and referrals.
- Consulting with industry analysts. Gartner's annual RIM market guide, and Gens & Associates World Class RIM report both provide an overview of RIM vendors. (Note, however, that both include both pharma and medtech-focused solutions within their respective guides.)
Build vendor short list
Based on the information gathered in the previous step, you should be able to create a short list of two to six vendors. This may require short conversations with prospective vendors, but you should have your short list before you schedule product demos and/or send out a request for proposal (RFP).
Tip: If you communicate with vendors that don’t make your short list, let them know so that they don’t continue to contact you!
Evaluate vendor capabilities
This part of the project varies greatly from company to company, but your process should ensure that all of your stated requirements are being evaluated against each vendor’s capabilities. Not all team members need to evaluate all requirements – individuals should be assigned based on their understanding of the area being evaluated. The same people, however, should evaluate the same requirements across all vendors to ensure a fair comparison.
If your organization requires an RFI (request for information) or RFP (request for proposal), those need to be compiled and sent to the vendors as the starting point for vendor evaluations. These documents allow your team to gather the same information from all vendors. Put simply, these are documents that list your requirements and ask the vendors to indicate if they address them natively within their software, through third-party integrations, or not at all. Our RIM Buyer’s Guide provides a template that can be used as a starting point.
Whatever your evaluation process looks like, your team needs to see the software. For systems as large as RIM solutions, you may need multiple demonstrations with the vendor and your team. Work with the vendor to determine how the process will work, but typically you will have an overview demonstration and then separately schedule individual sessions, if they are needed, to cover specific features or answer additional questions. While everyone on the evaluation team should attend the initial demo, additional sessions should be scheduled only when needed and only with those team members required. The following can help ensure a smooth process:
- Communicate clearly to your team what they are responsible for during the demo. Who is taking notes? Are different team members responsible for evaluating features in different areas of the software?
- Set expectations with the vendor ahead of time and maintain control of the demo agenda. The software vendor will know what sequence works best for their product, and you should allow them to guide you. However, you will want to steer them away from spending too much time on features that are not important to your team.
- Ask the vendor to keep track of unanswered questions or features that you were unable to see. The vendor should be expected to follow-up on these items.
Rate and rank vendors
Using your requirements list, each vendor should be rated for each requirement. Require vendors to clearly indicate if a requirement is met “out of the box,” requires custom development work, or is not supported at all. Consider a scale of 0-5, with 0 being a feature the vendor does not support. Multiplying the rank by the importance of the feature (3 – Critical to have, 2- Important to have, 1- Nice to have), will give you a good picture of where each vendor ranks.
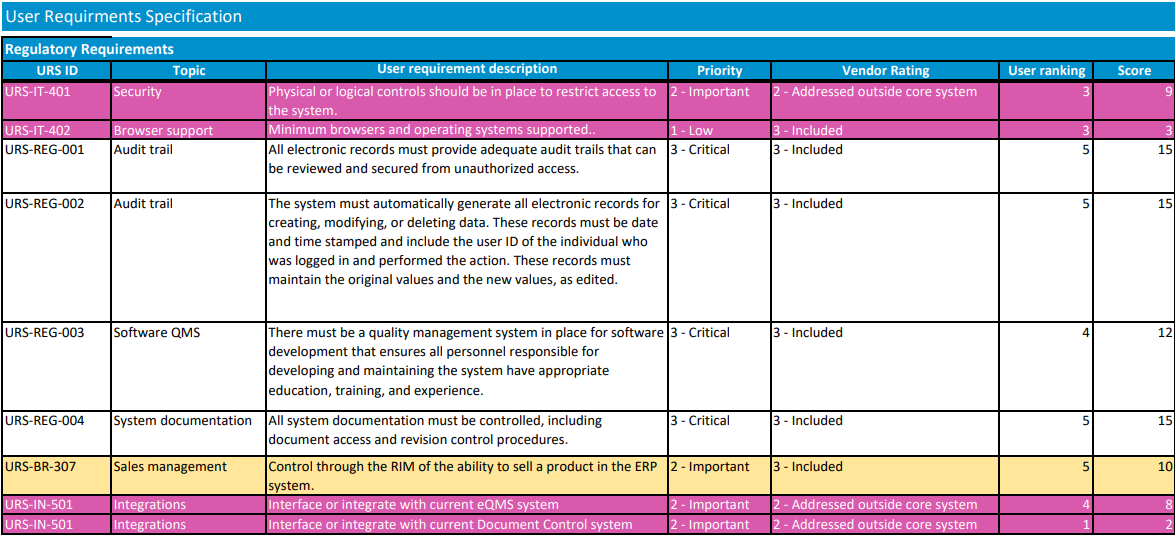
There should be some subjective items that are used in rating, also, such as how easy you believe the vendor will be to work with. Once the vendors are rated, the team should meet to discuss differences between team members' ratings and then to agree on where each vendor ranks. It is important that all requirements are considered and weighed appropriately while ranking vendors. For example, selecting the system with the best price may leave you with a vendor that doesn’t have the resources to support your implementation.
Negotiate and purchase
Hopefully, you will be able to successfully (and quickly) negotiate with your top-ranked vendor. However, it does sometimes happen that an agreement cannot be reached with the initial vendor for reasons that may include pricing adjustments or unexpected changes to the availability of their resources. In this case, you will need to move on to your second choice.
For more information on specific criteria for purchasing a RIM system, read our RIM Buyer’s Guide.


GET IN TOUCH






.avif)
