
What is software as a medical device (SaMD)?
As the pace of technological innovation continues to increase, the definition of what constitutes a medical device also continues to evolve as countries update regulations. In 2013, the International Medical Device Regulators Forum (IMDRF) created the Software as a Medical Device working group. Currently chaired by the U.S. FDA, the working group is chartered with developing guidance that encourages innovation while assuring safe and effective products. While SaMD is regulated differently in different countries, this article will focus on the many similarities, and some differences, between the FDA regulations in the U.S. and the MDR regulations in the EU.
In general, medical device software falls into 3 different categories:
- Software as a Medical Device (SaMD): The IMDRF defines SaMD as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” We list specific examples below, but typically the software classified as SaMD isdesigned to run on generally available hardware, such as Windows computers or mobile devices, or online in the “cloud”. While they may be utilizing data from another medical device, SaMD performs its function independently of any medical equipment or hardware.
- Software in a Medical Device: Sometimes referred to as SiMD, Software in a medical device cannot operate separately from its device, or perform its primary function without the device. For example, the software used to program and run an MRI machine would be useless without the MRI machine. Software in this category is regulated together, and as part of, the whole device.
- Software as an Accessory to a Medical Device: Similarly to Software in a Medical Device, software in this category cannot fulfill a medical purpose on their own. In some cases, a manufacturer may be able to bundle or embed the software in the medical device (making it SiMD) and/or also sell the software separately, making it an accessory.
Similarly, in Europe, the European Commission’s Medical Device Group (MDCG) defines Medical Device Software (MDSW) as a having its own intended purpose. Software which controls a medical device or is otherwise part of a medical device and does not serve a separate medical purpose does not qualify as MDSW, but is regulated by the MDR.
Software in the SaMD category is both a medical device AND software - with relevant regulatory and quality considerations that are specific and unique to each category, yet which must work in tandem. For example, software development best-practices, referenced by the IMDRF SaMD working group, call for iterative feedback loops allowing for quick turnaround of feature requests and bug fixes. While the ability to provide the latest technology and features to the market is an important advantage with SaMD, it does not supersede applicable medical device regulations governing patient safety and efficacy.
Examples of software as a medical device (SaMD)
Because SaMD software, by definition, is capable of running independently of any specific medical device or hardware, there is a relatively clear line between Software as a Medical Device and other medical software:
SaMD regulatory overview and framework
Why does SaMD have independent regulatory considerations?
Over the past decade, the IMDRF, FDA, and other regulatory bodies have worked to better align regulations with the quickly evolving capabilities and nature of digital devices. Software-only devices (SaMD) generate unique opportunities and considerations:
Because SaMD software typically runs on publicly available hardware:
- SaMD software must not only be designed to work on specific platforms (usually multiple), but must also be tested and updated frequently as new hardware and operating systems become available.
- Agile software development methodologies can provide an environment in which fast product feedback loops are supported within the required regulatory framework.
Because SaMD software can be made readily available to the general public, or specific patient groups, using their own devices:
- SaMD software can generate faster user/patient feedback - both for medical professionals and for the device manufacturer. Given this environment, regulatory bodies generally want to enable the market to safely take advantage of new innovations as quickly as possible.
- Product testing needs to take into account the unique and varied environments in which the software may be used, potentially in environments the product is not intended for. The software developer cannot control updates to operating systems or internet browsers, other software that may be running on a user’s device, or the ability for the user to potentially share software or data with others.
- The advantages of quickly delivering product fixes and updates need to be measured against potentially introducing new, possibly misunderstood or unwanted features, to all users at once.
SaMD and MDSW risk-based categories
As with all medical devices, the FDA and European MDR classify SaMD and MDSW, respectively, based on the potential impact to patient or public health. The SaMD categorization framework from the IMDRF is an effort “to introduce a foundational approach, harmonized vocabulary, and general and specific considerations, for manufacturers, regulators, and users alike to address the unique challenges associated with the use of SaMD...” This framework provides guidance only for today’s medical software developers, as well as regulatory bodies, such as the FDA. The chart below lists these risk categories by the state of the healthcare situation or condition and by the significance of the information provided by the SAMD:
SaMD categories
SaMD Category I:
SaMD Category I software can provide information for both Serious and Non-Serious health conditions or diseases. Software dealing with serious conditions can be classified in Category I only if it is providing information to inform, not drive, clinical management and is of low impact. Otherwise, Category I SaMD software provides information related to non-serious diseases or health conditions.
Example: Software that collects exercise-related data, such as heart rate, number of steps, and distance walked. If the information is stored and/or transmitted for use by a qualified professional the software is considered to be in Category I, as long as the information is not used by the software to make treatment recommendations directly to the patient or healthcare provider.
SaMD Category II:
SaMD software in Category II may be used to provide information relevant to a non-serious, serious, or critical healthcare condition or disease, depending on the significance of the information it provides to the healthcare decision. Software that is used to treat or diagnose a health condition is only classified in Category II for non-serious health conditions. Software that provides information regarding serious or critical health conditions or diseases will be classified in Category II only if it provides information to drive or inform, respectively, clinical management decisions (as opposed to providing treatment or diagnosis recommendations directly).
Example: SaMD that monitors a diabetic patient’s carbohydrate intake and blood glucose level to calculate recommended insulin dose.
SaMD Category III:
SaMD software that provides information to treat or diagnose serious conditions - or which drives clinical management for a critical condition - is classified in Category III.
Example: SaMD that analyzes individual data from at-risk populations for a specific type of cancer, and is used to develop preventative intervention strategies.
SaMD Category IV:
SaMD Category IV software provides information used to treat or diagnose a critical health condition and is considered to be very high impact.
Example: SaMD that evaluates images of skin lesions in order to determine malignancy.
For SaMD intended to be used in multiple healthcare situations, the software will be categorized at the highest category according to the SaMD definition statement.
SaMD within the FDA and MDR regulatory framework
The graph below relates SaMD categories with FDA medical device classes, and further identifies where an independent review is recommended by the FDA. For additional information see Software as a Medical Device (SaMD): Clinical Evaluation. Guidance for Industry and Food and Drug Administration Staff.
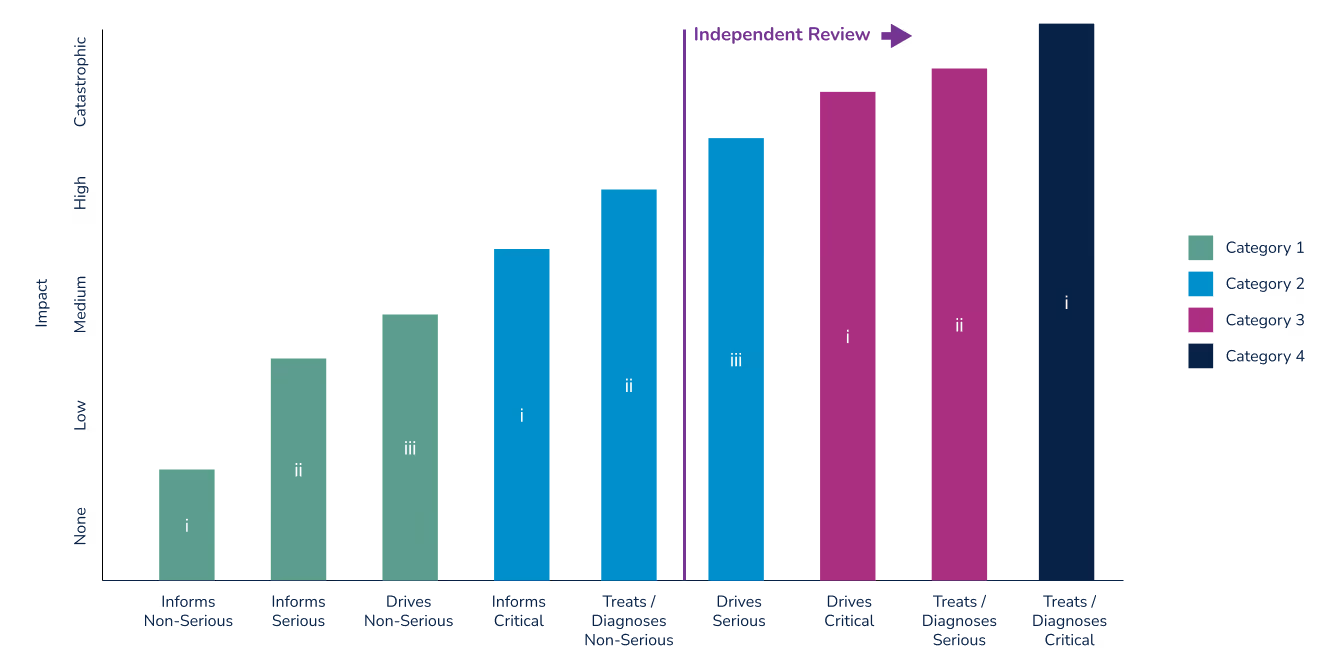
Note that FDA regulatory classifications more closely align with the IMDF’s SaMD categories than do those defined by under the European MDR. In the U.S., a device is classified by identifying similar, predicate devices. The simpler 510(k) pre-market submission can be used if a manufacturer can show that their device is substantially equivalent to a Class I or II device. With few exceptions, devices that are Class III are subject to the more rigorous Pre-Market Authorization (PMA) process. Devices where no substantial equivalent can be shown are subject to to the PMA or De Novo regulatory submissions processes. The De Novo process was implemented in 2010 to provide a pathway for novel devices with lower risk profiles.
Medical Device Software is classified by the European Commission’s Medical Device Coordination Group (MDCG) into Class I, II, or III. For devices falling under the IVDR, the MDR defines four classes using letters; A, B, C, and D.
In the EU, a rules-based framework is used to classify devices. MDSW classification is relatively complex, using a series of 22 “waterfalling” rules with yes/no questions that lead to a final classification of each device. While we won’t go into detail in this article on each of the rules, Rule 11 does deserve discussion. (You can read the Guidance on Classification of Software for MDR and IVDR here).
The MDR’s “rule 11” regarding MDSW classification was released in 2017. This rule states, in part, that “Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa.” The rule then provides 2 exceptions that increase the MDSW classification in cases where the decisions could cause “a serious deterioration of a person’s state of health or a surgical intervention” (class IIb), or could cause “death or an irreversible deterioration of a person’s state of health” (class III).
As a result of this rule, very few MDSW products can be classified as Class I, meaning that the majority of MDSW is subject to conformity assessments by a Notified Body. Medical software manufacturers marketing their product in Europe should therefore not rely on previous product classifications as a guide.
The future of regulatory approval for software as a medical device (SaMD)
The FDA recently released a new draft guidance document addressing the Content of Premarket Submissions for Device Software Functions. This is a major update of the 2005 guidance on pre-market submissions for software in medical devices and is also long overdue, given the pace of change in the software industry (the FDA originally committed to delivering this update in their fiscal year 2019 as part of the MDUFA IV agreement). This guidance sets out requirements for all pre-market submissions, including 510(k), DeNovo, and PMA submissions.
The recent draft document provides additional guidance around the documentation required for premarket submissions. It defines an “enhanced” level of documentation required for software that is a Class III device, combination product, Blood Establishment Computer software, or when the failure of the software would present probable risk of death or serious injury.
Staying on top of medical device classifications
Classifying a medical device and gaining regulatory clearance can be a complex process, especially if the device contains software or other emerging technology where some regulatory bodies may be further along than others in developing applicable regulations. For additional information about some of the common pathways to market for new products in the U.S., read our Beginner’s Guide to the FDA 510(k) and Beginner’s Guide to the FDA De Novo Classification Process.
Interested in learning how Rimsys can automate your submission process? Request a custom demo of our RIM platform.


GET IN TOUCH






.avif)
