
Blogs
MedTech
The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR)
This article is an excerpt from The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR) ebook.
Table of contents
- Overview
- Terminology
- EU MDR/IVDR Annex I
- EU MDR/IVDR Annex II
- Proactive Monitoring & Maintenance
- Comparison Table: EU MDR/IVDR Annex I GSPRs vs EU MDD/IVDD Annex I Essential Principles
With the initial rollout of the European Medical Device Regulation (MDR) complete, medical device companies are shifting focus to the sister In Vitro Diagnostic Regulation (IVDR) which has rolling effective dates starting in May 2022. Like the MDR, the IVDR also includes new General Safety and Performance Requirements (GSPR). The expanded 2nd edition of this ebook includes a detailed summary of the IVDR GSPR regulations in addition to those of the MDR. It provides you with practical guidance on how to meet the GSPR requirements for all types of medical technology products. This ebook, however, should not take the place of reviewing the actual regulations and consulting regulatory experts when needed
Timeline
The EU MDR submission became mandatory from the previous MDD directive on May 26, 2021, and the EU IVDR effective date is quickly approaching. In fact, all submissions for new devices under the new EU IVDR must be implemented no later than May 25, 2022. Below is a high-level overview of key dates for both regulations.
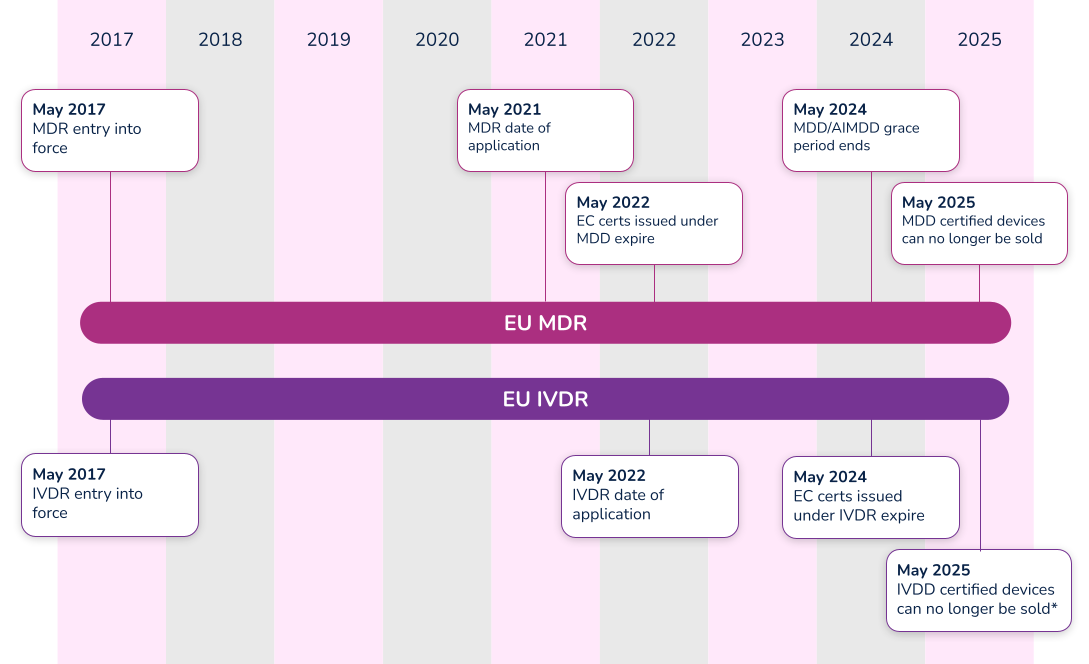
*Note that the timeline for compliance was extended in 2021. Class D (high-risk) devices have until 2025 to comply with IVDR, while Class C devices have until 2026. Class B and Class A sterile devices have until 2027 to comply with IVDR.
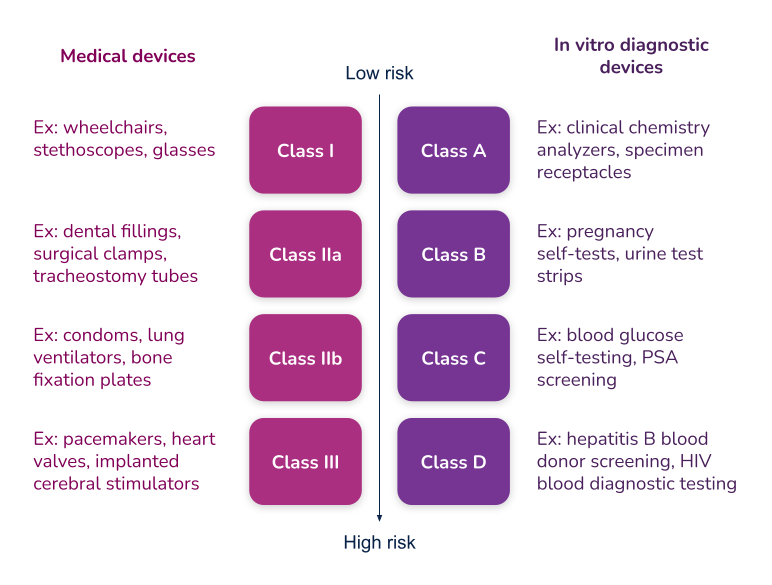
What’s the difference between Essential Requirements, General Safety and Performance Requirements (GSPR), and Essential Principles. In order to have a meaningful dialogue, let’s first discuss the three (3) main terms used in the industry.
#1 Essential requirements
The ‘Essential Requirements’ is the backbone for establishing conformity with the Medical Device Directive (MDD 93/42/EEC) and the Active Implantable Medical Device Directive (AIMDD 90/385/EEC). Detailed within Annex I of the MDD and AIMDD, the ‘Essential Requirements’ laid out the requirements that devices must meet in order to state compliance to the directives. With the implementation of the new EU Medical Device Regulation (MDR 2017/745), the ‘Essential Requirements’ will become superseded by the new EU MDR General Safety and Performance Requirements (GSPRs).
#2 Essential principles
The IMDRF laid out Essential Principles requirements in a document entitled Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices. From a high-level perspective, three basic tenets make up these ‘Essential Principles’:
- A device must be designed to be safe and perform effectively throughout its lifecycle.
- Device manufacturers must maintain all design characteristics.
- Devices must be used in a way that is consistent with how it was designed.
Many countries use the term ‘Essential Principles’ when compiling the documentation required to determine compliance to the law. For instance, the Australian Therapeutic Goods Administration (TGA) uses the term ‘Essential Principles Checklist’. Regardless of the term used, Essential Principles are of similar nature and overlap many of the Essential Requirements and new GSPRs.
#3 General safety and performance requirements (GSPR)
As of May 26, 2021, medical device manufacturers must start to comply with Annex I – General Safety and Performance Requirements (GSPRs) of the new EU Medical Device Regulation (MDR 2017/745). GSPRs are specific to the European MDR and IVDR. If you hear any other term (i.e. Essential Principles), it most likely means it is not referencing the European market.
Annex I of the EU MDR and IVDR details the specific requirements of the General Safety and Performance Requirements (GSPRs). The GSPRs are broken down into three (3) chapters in Annex I, MDR 2017/745 and IVDR 2017/746:
- Chapter 1 - General requirements
- Chapter 2 - Requirements regarding design and manufacture
- Chapter 3 - Requirements regarding the information supplied with the device
Chapter 1 - General requirements
Both the EU MDR and the EU IVDR outline General Safety and Performance Requirements (GSPRs) in great detail for medical device designers and manufacturers. The general requirements for each are almost identical and consist of the following:
- Devices must perform in a way that aligns with the intended design.
- They must not compromise the health or safety of a patient, user, or any other person associated with the device.
- Risks must be reduced as much as possible, but not so much that they negatively affect the risk-benefit ratio.
- Device manufacturers must implement and maintain a thorough, well-documented, and evaluative risk management system that continues to be updated throughout the life cycle of a device.
- Manufacturers and designers must include any necessary measures for protecting users in cases where risks cannot be completely eliminated.
- Manufacturers must provide users with information about any potential risks that remain. This information must be clear, easy to understand, and considerate of the users’ technical knowledge level, use environment, and any applicable medical conditions.
- Devices must withstand the stresses of normal use for the duration of their lifecycle. Devices must be designed, manufactured, and packaged in a way that protects them from damage during transport and storage.
- When it comes to risks and negative side effects that are known and foreseeable, designers and manufacturers must make every effort to minimize negative outcomes. They must also ensure that potential risks are acceptable when compared to the potential benefits of a device to its users.
Chapter 2 - Requirements regarding design and manufacture
The GSPRs also provide key details regarding specific information about the performance, design and manufacture of medical devices. As it relates to design inputs, the MDR and IVDR GSPRs provide highly detailed requirements relating to a device’s technical information. Further detail can be found in the comparison tables in Appendix A and Appendix B, where we have compared MDR to MDD and IVDR to IVDD.
Chapter 3 - Requirements regarding the information supplied with the device
The final key area of governance within the GSPRs relates to specific information a manufacturer must supply with a device. The general requirements for this information states that, “Each device shall be accompanied by the information needed to identify the device and its manufacturer, and by any safety and performance information relevant to the user, or any other person, as appropriate.” The requirements provide further detail as far as location - specific information that must be provided on the following:
- The device label includes its UDI.
- The user instructions.
- The packaging of a device that is intended to maintain its sterile condition.
Medical devices are subject to significant regulations and a full understanding of EU MDR and/or IVDR labeling as defined in Annex 1 Chapter 3.
In addition to the specific requirements identified within Annex I of the EU MDR and IVDR, Annex II, Technical Documentation, identifies additional requirements. Specifically, in both EU MDR and IVDR’s Section 4 – General Safety and Performance Requirements it states:
“the documentation shall contain information for the demonstration of conformity with the general safety and performance requirements set out in Annex I that are applicable to the device taking into account its intended purpose, and shall include a justification, validation and verification of the solutions adopted to meet those requirements. The demonstration of conformity shall include:
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
(c) the harmonised standards, CS or other solutions applied; and
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonised standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation.”
Let’s break this down into each part.
Requirement
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
What needs to be documented for the requirements that apply or the requirements that do not apply?
Each and every section of the EU MDR GSPR or EU IVDR should be assessed in its own right as it pertains to your medical device. When a requirement applies, a simple statement may be made that this requirement applies to the device. In practice this is often achieved using a checklist or table, with a column for applicability and a Yes/No answer against each requirement. When a requirement applies, you can move on to the other parts of demonstrating conformity regarding methods used and standards applied.
When a requirement is not applicable, a statement must be made to that effect, i.e. a ‘No’ in the applicability column. Additionally, it must be fully and properly justified. Such a justification may be something like ‘The device is not powered and is therefore not an active device. This requirement does not apply.' The justification should clearly state why the requirement has been deemed not to apply so that your notified body can understand your reasoning
Requirement
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
What is meant by “method or methods used”?
This relates to the way you complied with that GSPR requirement, historically it would be listed as a standard or other documentation reference that you have applied to demonstrate compliance, however, the question of ‘method or methods used’ is new to the MDR and it is expected that a verbal description be provided such as:
i. Risk analysis weighed against clinical evaluation benefit
ii. Performance intended demonstrated by design requirements, verification and validation
Requirement
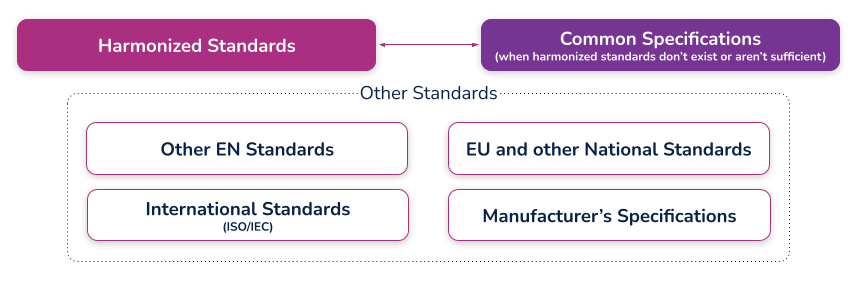
(c) the harmonized standards, common standards (CS) or other solutions applied;
What are harmonized standards, common specifications (CS), and “other solutions”?
Harmonized standards
These are standards that have been specifically developed and assessed for compliance to a regulation or directive. They are published in the Official Journal of the European Union (sometimes just referred to as ‘the OJ’) and if you comply with these standards then there is a ‘presumption of conformity’ with that directive or regulation to which they have been harmonized. These harmonized standards can only be created by a recognized European Standard Organization (such as CEN or CENELEC). When a standard is harmonized, an annex is added that describes how the standard conforms to the directive or regulation. When using harmonized standards, you should make sure that you understand how the standard conforms so that you do not claim compliance when the standard either does not meet that requirement or only partially meets that requirement.
If a standard does not meet a certain requirement of the directive or regulation, or indeed only partially meets it, then you must employ additional mechanisms for compliance. If a harmonized standard meets part of a directive or regulation, then by complying with that standard you also fully meet the corresponding requirement(s) The list of harmonized standards continues to grow - refer to the “Healthcare Engineering” section of the European Commission’s Harmonized Standards page for current information. In this case, using an MDD harmonized standard and documenting a justification for doing so (i.e. how you believe the standard demonstrates compliance with the GSPRs), should provide sufficient evidence
Common specifications
Common Specifications (CS) are a new concept in the MDR. They allow the European Union to add additional requirements that must be met in order to claim compliance where harmonized standards do not exist or where relevant standards are considered insufficient. The definition of a Common Specification is:
‘A set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.’
Requirement
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonized standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross- reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation;
What is the expectation for incorporating a "cross-reference to the location of such evidence within the full technical documentation"?
This means that someone looking at the document should be able to identify exactly where in the technical documentation that the compliance evidence can be found. For example, this may refer to test reports and their exact location, or it could even reference locations within a large document, depending on the GSPR and your particular documentation. (i.e. if you have included usability risks as part of a larger risk assessment, you may need to say ‘See Technical File XXX, Section XX, Doc RMF001 rev 3 lines 65-78’). In other cases it could just mean the whole document reference, i.e. Have you done risk management? – then yes, it is RMF001 rev 3. What the specific reference actually is depends on how you have managed your technical documentation and how defined it is (i.e. separate reports or one big one). There should be no ambiguity as to where the document is located
An example of a completed GSPR checklist could look something like this (applicable and nonapplicable examples are shown):
Specification developers and manufacturers must continually maintain their technical documentation to stay compliant. Part of this process is to ensure that they take into account the "generally acknowledged state of the art".
Proactive monitoring
'State of the art'
There is no formal definition of ‘state of the art’ within the EU MDR or IVDR, although it is mentioned many times. ‘State of the art’ is an ongoing debate; however, it generally means that it embodies what is currently and generally accepted as good practice in the medtech industry. The ‘state of the art’ does not necessarily imply the most technologically advanced solution.
One consensus on state of the art is being up to date and compliant with the current and in effect standards that are applicable to your device. This means that if a standard is updated that your medical device is compliant with, you must evaluate that update to ensure that it would meet the EU MDR or EU IVDR ‘state of the art’ requirement. This is not a new requirement from the EU MDD but it is spelled out more clearly in the EU MDR.
The specification developer or manufacturer is ultimately responsible for determining if the updated standard applies or does not apply to their device(s). Either way, the justification should be documented within a gap analysis.
Monitoring for changes
Of course, 'state of the art' only applies if you actually know if something changed. This is why you need to develop a process for monitoring the standards that compliance is claimed. Every single standard that is associated with your technical documentation must be actively monitored, reviewed, and reported on.
If you have a product on the market and need a better way to monitor and maintain your General Safety and Performance Requirements (GSPR) or Essential Principles, Rimsys can help. Rimsys digitizes and automates GSPR and Essential Requirements so you can dynamically update and proactively monitor changing standards and evidence files.
When a standard or evidence file changes, you will automatically be notified and can update one GSPR or all of your GSPRs as applicable with a single click of a button. If additional information is needed, such as testing, it’s also invaluable to ensure that all devices are identified. What used to take weeks of manual, error-prone administrative tasks is now done in seconds within a fully validated, secure, maintenance-free, cloud-based solution
Maintenance
Maintaining and updating your technical documentation is generally the hardest part of staying compliant. Robust processes must be established to ensure nothing slips through the cracks and show up as nonconformances during regulatory audits.
Gap analysis
In addition to meeting the ‘state of the art’ requirements and the continuous proactive monitoring of standards, once a change has been detected that affects the technical documentation, a proper and thorough gap analysis must be completed.
The gap analysis between the old versions and the new versions, or an evaluation of a brand new standard, must occur and be properly documented. The gap analysis should detail what is applicable and what is not applicable, with your supporting justification.
If something within the new or revised standard was applicable to your device, additional engineering testing, documentation, justification, and, in some instances design changes, may be needed to ensure compliance
GSPR updates
Once the gap analysis has been properly documented, specification developers and manufacturers must update their GSPRs.
These updates include finding the withdrawn or superseded standard or evidence file throughout each row within your GSPR table, for every single device on the market on which this change is applicable. This could be one table or dozens of tables depending on the complexity of the products and your product mix.
Without a holistic RIM system to help you, this is an error-prone process as is it tedious, administrative, and extremely easy to miss an inappropriate referenced standard or evidence file.
Extreme diligence on the regulatory or engineering team must occur to ensure these critical updates to the GSPRs are not missed and a gap analysis must be properly referenced throughout. Any justification for including or excluding a new standard or evidence file will be scrutinized by regulatory auditors, and without proper maintenance, may lead to additional review time.
To continue reading this eBook including Comparison Table of the EU MDR Annex I GSPR vs. the EU MDD Annex I Essential Requirements, please register to download the full version.


GET IN TOUCH






.avif)
