


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.
.avif)

2023 Regulatory performance report
Today at Rimsys, we unveiled the 2023 MedTech Regulatory Performance Report, a new set of insights into the state of medtech regulatory affairs. Compiled based on interviews with 200 regulatory professionals and executives, the study provides a detailed look into how regulatory teams are staffed, their processes, the tools they use, and ultimately how they perform.
Why did we create this study? There were two driving factors behind the research. The first was a common theme that we heard from a number of our customers: Regulatory leaders don’t have clear data and benchmarks. They don’t necessarily know how long a new market submission should take, and how to plan for or assess the work of their teams. While other studies look at the medtech industry broadly or the state of the regulatory profession, this study tries to build a comprehensive resource for regulatory (and company) leaders.
The second factor was really for ourselves and the team at Rimsys. As a company building solutions specifically for medtech regulatory affairs, we wanted more insight into where companies were successful, where they struggled, and where we can add value.
What did we find? Regulatory teams perform a lot of hero work and rate themselves highly for their accomplishments. At the same time there is a lot of opportunity for process improvements, and companies that invest in digital transformation for regulatory affairs see better performance.
Regulatory professionals are superheroes
Regulatory teams are generally pretty small. Most companies have less than 10 full-time regulatory professionals. These small teams complete an enormous amount of work. Last year on average, RA teams completed 50 license renewals, 50 license updates, and 10 new market submissions. This is impressive output.
Digging a bit under the covers, we found that this output relied heavily on the support of external consultants. 90% of companies use consultants to keep pace with their regulatory workload. Front-line employees also struggle with burnout. They were much more likely to report feeling under-resourced than regulatory leaders.
But process problems persist
A lot of regulatory work remains extremely manual. 70% of regulatory teams spend half their time or more on repetitive administrative tasks. All of this manual work increases the frequency of errors and required rework. 61% of companies reported a major non-compliance incident in the past 2 years.
Manual work also makes it difficult to complete regulatory projects in a timely fashion. Teams completed a lot of projects, but each took a long time. Over half of all companies spend 4 months or more on license renewals, license updates, and new market submissions.
Moving regulatory affairs forward
As regulatory requirements become more complex, there’s a natural question about how teams will work moving forward. MDR & IVDR in Europe have significantly increased the regulatory workload required to bring and keep products on the market. Will organizations be able to keep pace with the same resources, tools, and processes?
No, and the performance report shows that medtech companies are investing to improve their regulatory capabilities. The majority of companies are planning to increase the sizes of their RA teams in 2023, and 40% expect to increase their investments in regulatory software. Companies are increasingly adopting specialized software to better support regulatory processes.
Dig into the survey results
The full survey results provide insights into more aspects of regulatory performance. They show that companies need to take a deeper look into their processes and how regulatory resources are allocated. There are two ways to learn more:
- Visit the survey page to see the full results (the survey whitepaper can be downloaded at no cost)
- Watch the recording of our webinar with PA Consulting. We discuss the survey results in more detail and share our regulatory predictions for 2023



Post-market surveillance for medical devices in the European Union
This article is an excerpt from Post-market surveillance for medical device in the European Union.
Table of Contents
- What is post-market surveillance?
- What classes of medical devices require post-market surveillance?
- Components of a successful post-market surveillance plan
- PMS data requirements
- Post-market surveillance system goals
- Required post-market surveillance reporting
- Embracing post-market surveillance as an integral part of your quality program
- Getting started with post-market surveillance
Post-market surveillance (PMS) is designed to monitor the performance of a marketed medical device by collecting and analyzing field use data. Article 10 of the EU MDR and IVDR requires all device manufacturers to have a post-market surveillance system in place. The main elements of the PMS are laid out in Article 83, and additional details for lower-risk and higher-risk devices are covered in articles 84 and85, respectively.
In general, a PMS system consists of both proactive activities and reactive, or vigilance, activities. While post-market surveillance and vigilance are sometimes used interchangeably, vigilance consists of separate activities that feed post-market surveillance programs.
Post-market surveillance systems are used to collect and analyze data not only about the manufacturer’s device but also about related competitors’ devices that are on the market. Data collected through PMS procedures is then used to identify trends that may lead to, among other things, quality improvements, updates to user training and instructions for use, and identification of manufacturing issues.
Note that “market surveillance” encompasses activities performed by a Competent Authority to verify MDR compliance, and should not be confused with the topic of this ebook,“post-market surveillance," which is performed by the manufacturer.
All medical devices marketed in the EU require some level of post-market surveillance, and all medical device manufacturers must implement a post-market surveillance system (PMS). The requirements of the PMS, however, vary and should be “proportionate to the risk class and appropriate for the type of device” (MDR Chapter VII). In particular, the type and frequency of reporting vary based on a device’s risk class.
A post-market surveillance plan (PMS) is an integral part of a manufacturer’s quality management system and provides a system for compiling and analyzing data that is relevant to product quality, performance, and safety throughout the entire lifetime of a device. The PMS should also provide methods for determining the need for and implementing any preventative and corrective actions. A PMS system should include and define:
Surveillance data sources
With the increased focus on proactive risk identification in the MDR, it is important to design post-market surveillance systems that actively acquire knowledge and detect potential risks. It is not sufficient to rely solely on spontaneous reporting by healthcare providers, patients, and other stakeholders.
In addition to information coming from Clinical Evaluation Reports and complaint and adverse event reporting, typical sources of surveillance data include:
• Social media networks: Because many of your stakeholders may be communicating on social media networks, it is important to employ social listening techniques and/or tools to identify issues and concerning trends as they develop.
• Industry and academic literature: Any studies, academic papers, and other literature that addresses similar devices or the specific use cases for which your device is designed should be evaluated. In particular, risk factors and adverse events identified with similar devices should be closely examined. It is also important to identify newer technologies that may affect the benefit-risk ratio and establish a new definition of “state of the art” for the device type.
• EUDAMED: While the European Database on Medical Devices (EUDAMED) is not yet fully functional, it is intended to provide a living picture of the lifecycle of all medical devices marketed in the EU. Manufacturers should take special care to consider information for similar devices made available through the EUDAMED system in the future.
• Registries: Patient, disease, and device registries can provide information that informs the clinical evaluation process which provides input into the post-market surveillance system.
Data analysis methodology
A well-defined data analysis methodology will accurately identify trends and lead to defendable decisions in the application of post-market experience. Once the necessary information has been identified and collected, and potentially cleaned of incomplete or otherwise unusable data, the data needs to be analyzed.
The goal is to identify meaningful trends, correlations, variations, and patterns that can lead to improvements in the safety and efficacy of the device. There are many data analysis tools available that can assist with:
• Regression analysis that will identify correlations between data (e.g. the device location/geography correlates to battery life).
• Data visualization that can be useful in spotting trends in the data.
• Predictive analytics, which can be particularly useful with large data sets, to identify future trends based on historical data.
• Data mining, which is also normally used with large datasets, to organize data and identify data groups for further analysis.
Benefit-risk indicators and thresholds
The MDR requires that medical device manufacturers not only demonstrate the clinical benefit of their device but also quantify the benefit-risk ratio. The benefit of a device must be shown to clearly outweigh the risk for it to gain market approval. Article 2 (24) of the MDR defines the benefit-risk determination as “the analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose when used in accordance with the intended purpose given by the manufacturer.”
A PMS system should clearly define benefit-risk calculations and the data used to support them. Post-market surveillance activities are critical in order to re-evaluate and maintain the benefit-risk calculations and determinations of a device throughout its life. Information that is gained through a PMS system can lead to:
• Identification of new risk factors.
• Adjustments to risk frequency and/or severity values based on actual use data.
• Adjustments to established risk calculations based on new “state of the art” technologies becoming available.
• Adjustments to established benefit calculations based on actual use data.
While complaint handling and other feedback tracking are more often described as part of post-market vigilance systems, they play a role in the more proactive post-market surveillance processes as well. A PMS system should define ...
To continue reading this ebook, download the full version.
.avif)
Rimsys Enters Strategic Alliance Relationship with KPMG
PITTSBURGH – March 11, 2025 -- Rimsys, the global leader of MedTech Regulatory Information Management (RIM) software, today announced that it has entered into a strategic alliance relationship with KPMG to advance digital transformation in the MedTech industry.
“KPMG’s deep experience in advisory and business transformation services and exceptional reputation make them a valuable alliance relationship for us,” said James Gianoutsos, Founder and CEO of Rimsys. “KPMG is on the cutting edge of industry trends and has a wide breadth of experience in helping companies innovate and scale. We are thrilled to work with them to help MedTech teams transform their regulatory management processes and leverage the benefits of automation and digitization as part of their broader transformation strategy.”
Founded for and by MedTech regulatory affairs professionals in 2017, Rimsys was created to bring efficiency to regulatory information management and fill an inhibitive technology gap in an underserved industry. Rimsys has since grown to support the world’s MedTech leaders backed by a staff that understands their complex workflows and a robust, secure technology infrastructure that allows customers to scale Rimsys software to support their changing regulatory needs and requirements.
“There is tremendous innovation happening in the MedTech industry, and we are excited to work with Rimsys to help clients transform how they manage regulatory information for getting new products to market and sustaining their existing product portfolios. It’s critical to approach these programs as a holistic business transformation across people, process, technology, data, and governance & controls,” said Dipan Karumsi, Principal, Consulting Sector Leader for Life Sciences at KPMG.
“Through our strategic alliance with KPMG, we can further expand our reach to large and enterprise MedTech companies and continue our exponential growth,” said James. “Combined with KPMG’s experience helping organizations mature their data collection and transformation processes to reach RIM readiness, we can enable the MedTech industry to innovate faster, strengthen compliance, and most importantly, improve the availability of life-changing medical technologies.”
See the full press release here.

FDA’s Final Rule on LDTs: What manufacturers need to know
In July 2024, the FDA's final rule in 21 CFR Part 809 on laboratory developed tests (LDTs) went into effect, amending its previous regulations to make it clear that IVDs, including those that are manufactured in laboratories, are classified as devices under the Federal Food, Drug, and Cosmetic Act. Our blog post provides an overview of LDTs, FDA’s final rule, the phase out policy schedule, and how LDT manufacturers can prepare themselves for compliance.
What are LDTs?
Simply put, LDTs are IVDs that are designed, manufactured, and utilized within a certified laboratory and are typically used for high-complexity testing.
Historically, FDA has used enforcement discretion only on LDTs, which means that most LDTs haven’t been subjected to specific regulatory requirements. However, the volume of and risks associated with LDTs have grown over the years. Some examples of modern LDTs include glucose tests, genetic tests for cancer and infectious diseases, and newborn screenings for early diagnostics. Without a regulatory framework in place, patients are at greater risk of receiving inaccurate test results, forgoing necessary or undergoing unnecessary treatment, and adhering to misleading or false product claims, possibly endangering patients and leading to higher healthcare costs.
FDA’s Final Rule on LDTs
Following the final rule that was issued, LDTs are now subject to the same regulatory requirements as other IVDs, including premarket reviews, quality system requirements, labeling requirements, adverse event reporting, and device listing and registration. To prevent disruptions in patient care, there is a four-year transition or phaseout period consisting of the following five stages:
- Stage 1 (May 6, 2025): LDT manufacturers will be expected to comply with FDA medical device reporting (MDR) requirements, correction and removal reporting requirements, and quality system (QS) requirements for complaint files.
- Stage 2 (May 6, 2026): LDT manufacturers will be required to comply with IVD registration and listing requirements, labeling requirements, and investigational use requirements.
- Stage 3 (May 6, 2027): LDT manufacturers will need to comply with all other QS requirements not covered in Stage 1.
- Stage 4 (November 6, 2027): Unless a premarket submission is received before the start of this stage, LDT manufacturers of high-risk products will need to comply with premarket review requirements for IVDs that may be classified into class III or that meet the requirements of section 351 of the Public Health Service Act.
- Stage 5 (May 6, 2028): LDT manufacturers of moderate and low –risk products will need to comply with premarket review requirements for IVDs unless a submission is received before the beginning of this stage.
Manufacturers of LDTs that don’t meet the requirements in each stage are deemed non-compliant to the regulations governing IVDs and may be subject to FDA 483 observations or warning letters, financial penalties, and even worse, involuntary removal of products from the market.
Note that some LDTs will be exempt from these requirements. Refer to the FDA’s website for more guidance.
Preparing for Compliance
Despite a four-year phaseout period, it’s crucial for LDT manufacturers to start assembling a compliance plan. Starting as early as May 2025, manufacturers will be required to comply with FDA Medical Device Reporting (MDR) requirements, correction and removal reporting requirements, and quality system requirements for complaint files.
It's good practice to conduct an internal regulatory assessment to ensure you have the resources, processes, and tools in place to successfully meet new requirements for LDT devices. It’s also essential to make sure your team is well-versed in these new requirements and the documentation and timelines involved. Including all relevant stakeholders early on, getting a comprehensive project plan in place, and meeting regularly to ensure all tasks are completed would be helpful during the phaseout period and beyond.
Regarding the LDT changes, the FDA has provided a Q&A sheet that you may find helpful.
If you're looking for guidance on FDA premarket submissions, see our Beginner’s Guides to the FDA 510(k), De Novo, and PMA processes.
How Regulatory Tools Can Help
FDA’s final rule on LDTs will add complexity to the regulatory information management of laboratory diagnostic tests. There are digital solutions that can help manufacturers stay current on updated regulations and manage the additional information and documentation needed because of these updates.
A regulatory intelligence database like Rimsys Intel can provide detailed global market entrance requirements, application timelines, fees, risk class specifications, and documentation needed for medical devices and IVDs so that manufacturers can start preparing their premarket strategies.
Regulatory Information Management (RIM) software like Rimsys can help boost efficiency, reduce compliance risk, and increase collaboration by centralizing regulatory information and automating time-consuming, manual processes. As a result, medical device manufacturers gain complete visibility into their submission management and selling status so that they can plan more effectively, avoid costly product delays, and execute faster.
%2520(1755%2520x%2520550%2520px)%2520(855%2520x%2520268%2520px)%2520(1).avif)
Planning Your Enterprise’s UDI Strategy for EUDAMED and Beyond
Rimsys recently hosted a webinar with RAPS titled “Planning Your Enterprise’s UDI Strategy for EUDAMED and Beyond.” During the session, our experts, Adam Price, Director of Regulatory and Technical Programs, and James Gianoutsos, Founder and CEO, provided best practices to help MedTech companies navigate the complexities of Unique Device Identification (UDI) compliance. The session covered key topics such as comparing US and EU UDI regulations, preparing for the January 2026 mandatory EUDAMED submission date, and using EUDAMED as a foundation for a global UDI strategy that can scale as additional markets adopt UDI requirements.
Here are some of the key topics we discussed to help Medtech teams prepare their strategy:
1. The importance of UDI to regulatory affairs teams
UDI is a globally unique identifier for medical devices that helps improve traceability, manage adverse event reporting, and ensure compliance with regulatory requirements. Regulatory affairs teams play a critical role in both establishing and maintaining UDI data to meet submission timelines and market placement requirements.
Responsibility for UDI management can vary between organizations, with some MedTech companies assigning the responsibility to IT or supply chain teams. When we asked our audience who is responsible for UDI management at their company, 80% of respondents said that it was their regulatory affairs team, emphasizing UDI’s crucial role in market placement.
2. Comparing US and EU UDI Requirements
FDA GUDID requirements have been in place since 2014. As such, some MedTech teams may plan to use their GUDID submission strategy as a baseline for the development of their EUDAMED submission strategy. While the US FDA’s GUDID system has standardized requirements, the EU’s EUDAMED is more complex due to additional data fields, language-specific entries, and the use of the EMDN nomenclature instead of GMDN. EUDAMED also introduces the Basic UDI-DI, which connects related devices within family groupings and links different EUDAMED modules. Companies must adapt their UDI strategies to account for these variations.
“Unlike the standardized US GUDID, the EU’s EUDAMED is more complex, featuring multiple device categories, language-specific data, and the EMDN nomenclature.” - Adam Price
3. The Interconnectivity of EUDAMED
EUDAMED consists of multiple interconnected modules. The Basic UDI serves as the key data element linking these modules together. This interconnected structure ensures that regulatory bodies, manufacturers, and economic operators can efficiently track devices throughout their lifecycle. Properly aligning UDI submissions with these modules is essential for seamless compliance and data consistency.
“The interconnectivity of EUDAMED is really important because the Basic UDI family groupings are the key piece of data that is going to tie the different modules together. For example, your notified body certificates for the regulations will have your Basic UDI numbers indicated on them. And then that's connected to the Basic UDI numbers that you have associated to your single registration number.” - Adam Price
4. Deadlines, Compliance Risks, and Recommended Actions per Device Category
The deadline for mandatory UDI submissions to EUDAMED is January 1, 2026, with a six-month grace period until June 2026 for devices already on the market. Failure to meet these deadlines could result in compliance risks, including regulatory penalties and restricted market access. Different categories of devices are subject to specific rules:
- Regulated Devices: Devices compliant with MDR or IVDR regulations must be linked to Basic UDI groupings and classified accurately under EMDN. Recommended actions include ensuring all data aligns with the new requirements and preparing for immediate submissions of new devices after January 2026.
- Legacy Devices: Devices compliant with older EU directives that remain on the market do not require a Basic UDI but must still have all essential data submitted. Companies should consolidate and verify legacy data, ensuring consistency with prior regulatory approvals.
- Non-Registered Devices: Devices no longer placed on the market but still have post-market surveillance activities must be registered for traceability. Manufacturers should identify these devices, gather historical data, and ensure that post-market surveillance records are aligned with regulatory expectations
.
5. Data Preparation and Submission Process
Data preparation for EUDAMED compliance is going to take some time, and the time to start preparing is now. It's important to recognize some of these high-level steps needed for effective UDI data preparation and submissions so that MedTech teams can plan appropriately:
- Identify: Collect and review all required UDI data, including language-specific fields and Basic UDI groupings.
- Verify: Ensure data accuracy and alignment with regulatory requirements, using EUDAMED’s business rules as a reference.
- Format: Structure data to meet EUDAMED’s formatting and enumeration rules, ensuring compatibility with submission methods.
- Submit: Transmit data using manual entry, XML uploads, or automated machine-to-machine (M2M) transmission, depending on the volume and complexity of your product portfolio.
“Ultimately, manufacturers need to be in a position to have all the data in for all products - not just the new products on the market but for all products by the end of that transitional period. It sounds a little apocalyptic but given the right planning, I think it's very achievable." - Adam Price
6. Getting Data into EUDAMED
Manufacturers have multiple options for submitting UDI data into EUDAMED, depending on their resources and portfolio size. Data can be manually entered through the EUDAMED user interface, uploaded via XML files, or submitted using automated machine-to-machine (M2M) transmission. Organizations managing large product portfolios are encouraged to adopt M2M transmission to ensure efficiency and data accuracy. Testing data submissions in the EUDAMED playground before official submission is highly recommended to identify and resolve any formatting or compliance issues.
“With more products, more data, and more BUDI families, there's more complexity. And as you're looking across your product portfolio, that's something that you need to consider as you plan your submission strategy.” -Adam Price
7. UDI Management Solutions
Organizations can choose from various UDI management solutions depending on their size and needs:
- Spreadsheets: Might be suitable for small companies with limited product portfolios but are time-consuming to manage and error-prone
- Repurposed Internal Systems: PLM or PIM systems can manage UDI data but may require manual processes and in-house expertise.
- Purpose-Built Connectors: Provide transmission capabilities for UDI data but don’t provide data capture and management
- RIM Systems (e.g., Rimsys): Provide a centralized, automated solution that integrates UDI data with product registrations and certificates, supporting seamless M2M transmissions.
“For large, complex product portfolios, RIM systems like Rimsys provide automated UDI management and seamless M2M transmissions.”
“Having an integrated data hub that's the source of truth for your UDI information is key. Then being able to associate your UDI data with all other data needed for UDI transmission in a RIM system, your registrations, certificates, legal entity, manufacturer, manufacturing locations, all the product data, all your BUDI data, having that all centralized in a RIM system is really critical to understanding and organizing your data in a way that can be meaningfully managed moving forward.” - James Gianoutsos
8. EUDAMED as a Foundation for Global UDI Compliance
There is a tremendous opportunity for EUDAMED’s comprehensive data requirements to serve as a foundation for global UDI compliance, as many countries are adopting similar regulations. Australia, Switzerland, the UK, and India are among the markets expected to enforce UDI requirements soon, with varying deadlines and data requirements. By using their EUDAMED data output as the foundation for a global UDI program, MedTech companies can build a UDI strategy that positions them to meet current and future requirements as they evolve.
“The application of UDI is not a one and done approach like it had possibly felt for manufacturers in the past. These requirements are going to need to be continually maintained from the perspective of your product characteristic data as well as new technologies as they're being introduced. So it's maintaining compliance to the current requirements, establishing compliance to the new countries’ UDI programs as those get rolled out, and being in a good position to control and consistently apply your UDI data.” -Adam Price
Conclusion
With the January 2026 deadline approaching, companies must act now to ensure compliance, streamline submissions, and prepare for future UDI requirements worldwide. Establishing a robust UDI strategy today will not only support compliance with EUDAMED but also lay the groundwork for expanding regulatory demands in other global markets. Leveraging centralized systems like RIM platforms can help manage the increasing complexity of global UDI regulations, ensuring consistent and efficient compliance.
“This seems to be a trend globally that UDI has become this single source data point to control and manage and maintain your products on the market. And so EUDAMED is just really the tip of the iceberg. Even though we know how complex it is, this is really just the beginning of this global UDI strategy that you have to be thinking about as you go into EUDAMED preparation.” -James Gianoutsos
Those interested in watching the webinar can access the recording here.
With an integrated and automated approach to UDI, Rimsys is a trusted and proven solution to help MedTech teams simplify EUDAMED compliance and assemble a global UDI program that meets current and future requirements. Request a custom demo rimsys.io/demo.

Quick reference guide - global medical device UDI requirements and timelines
This article was last updated on February 10, 2025.
What is UDI?
UDI systems are intended to benefit healthcare providers, manufacturers, authorized health authorities, hospitals and institutions, and individual consumers by providing:
- Faster discovery of possible flawed medical device information by health authorities.
- Quicker access to recall information, and visibility into current inventory.
- A reduction in medical errors through consistently documented product expiration dates.
- Identification of any counterfeit products being used in healthcare facilities.
- Assurances that information regarding an implanted device is safely retained and traceable.
UDI timelines and deadlines vary by market, classification risk, and product and have been revised multiple times in some countries*. This article details the UDI deadlines for the countries which have announced specific programs (draft or implemented) and is current as of the date of this article.
*Note: these dates can change as participating countries adjust their plans. We do our best to update this as more information becomes available.
Quick Links to country-specific sections:
- Australia UDI
- Brazil UDI
- Canada UDI
- China UDI
- European Union UDI
- India UDI
- Japan UDI
- Saudi Arabia UDI
- Singapore UDI
- South Korea UDI
- Taiwan UDI
- United States UDI
- UDI databases by country
General UDI labeling requirements
There are two components to a medical device UDI: the UDI device identifier (UDI-DI) and the UDI production identifier (UDI-PI). The UDI is presented as a barcode label (human and machine readable) on device packaging or on the device itself and acts as the access key to all device UDI attributes.
UDI-DI: This is the static portion of the UDI which identifies the manufacturer along with the specific device version. The UDI-DI (device identifier), also known as the Global Trade Item Number (GTIN) is assigned by an approved organization, such as GS1, and contains:
- Company prefix
- Manufacturers internal product code
- Check character
The UDI-DI is the primary identifier to be used in looking up device attributes in country-specific databases and is assigned prior to placing a product on the market. Note that the device identifier is different for different packaging levels of the same device.
UDI-PI: This is the dynamic portion of the UDI which is assigned by the manufacturer and identifies one or more of the following:
- Manufacturer’s lot or batch number
- Serial number
- Manufacturing date
- Expiration date
- Other attributes as defined by country-specific regulations
The UDI-PI actual values do not appear in country-specific databases (with the exception of the EU vigilance database).
Australian UDIGuidelines
Reporting Database: AusUDID (pre-production)
The Australian government for medical devices, the TGA, has not launched any official regulations or timeline for mandatory UDI labeling. They do provide a wealth of information on their website that is worth reviewing. In the meantime, however, they are hoping for a Q1 2025 implementation. The AusUDID Pre-Production environment is available for sponsors and manufacturers of medical devices supplied in Australia. It is a test environment that allows testing of data submission, prior to submission to the AusUDID Production environment. Any sponsor or manufacturer with an active TBS account can access the database.
ANVISA UDI guidelines
Reporting database:TBD
RDC No. 591/2021 is the regulations guideline for the identification of medical devices regulated by ANVISA, implementing the Unique Identification of Medical Devices (UDI) system. In July 2024, ANVISA finalized amendment RDC No. 884/2024 which implemented various adjustments to RDC 591/2021. The biggest take-away regarding UDI is the extension of one year on the implementation deadlines.
Health Canada website
Reporting Database: N/A
Position paper on the current state of UDI implementation
Medtech Canada strongly supports the global initiative led by regulators under the guidance of the International Medical Devices Regulators Forum (IMDRF), which aims to standardize the identification of medical devices by requiring that certain medical devices carry an internationally recognized UDI. Currently, there is no process in place for UDI in Canada.
China (NMPA) website
Reporting Database: China National UDI Database
Announcement No 22 of 2023
On January 1, 2021, the NMPA implemented the UDI system for its first batch of medical devices, including 69 Class III devices. The following year, June 1, 2022, followed the implementation for the second batch of other Class III medical devices (including IVD reagents). Then in 2023, Order No. 22 announced the third batch of products to adopt the UDI system.
As of June 1, 2024, medical devices listed in the third batch implementation product catalog must have already had UDI implemented. According to the degree of risk and regulatory needs, some Class II medical devices in the third batch included high-demand single-use products, items selected for centralized procurement, and medical aesthetic products, totaling 103 types in 15 categories.
European Union UDI Information
Reporting Database: EUDAMED
Rimsys Updated EUDAMED Timeline Blog Post
The UDI & Devices module is expected to be declared fully functional by the end of Q2 2025 and mandatory for industry use on January 1, 2026. The EU continues to strongly recommend to the industry to establish its solution and to submit data on a voluntary basis.
Medical Devices Rules, 2017
Legal Metrology Act, 2009
Reporting Database: N/A
Rule 46 of Medical Device Rule 2017 was set to require UDI labeling by January 1, 2022. However, details on how the UDI needs to be implemented have not yet been released but India's labeling and traceability requirements must be met as per CDSCO regulations.
In addition to the Medical Device Rule 2017, the Legal Metrology Act, 2009 focuses on standardizing weights and measures and ensures that packaged commodities, including medical devices, are labeled with accurate and clear information.
Law to Ensure Quality, Efficacy and Safety of Pharmaceuticals, Medical Device, and Similar Products
Reporting Database: N/A
There are two regulatory authorities responsible for regulation of medical devices in Japan: The Ministry of Health, Labour and Welfare (MHLW) and the Pharmaceuticals and Medical Devices Agency (PMDA). The MHLW is responsible for the administrative actions such as guidance and approval, and judgment on whether or not a product is considered a medical device. The PMDA undertakes product review and post-market safety measures.
As of Dec 2022, bar code labeling based on international standards is required for immediate containers/wrappings/retail packages of medical devices. It is expected for barcodes to be displayed on every medical device in unit of use for patients. Japan was an early promoter of standardized barcodes and is still working towards harmonizing the requirements with global UDI expectations.
The Pharmaceuticals and Medical Devices Act (PMD Act) translates in Japanese meaning "Law to Ensure Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, and Similar Products," but is often shortened to Act on Pharmaceuticals and Medical Devices or just PMD Act.
Requirements for Unique Device Identification (UDI for Medical Devices)
Reporting Database: Saudi-DI
The SFDA requires compliance with the Unique Device Identification (UDI) regulations on all medical device companies in Saudi Arabia for all classifications. Medical device classifications include: devices, IVD, non-medical IVD, chemical for medical use, distillation device, general lab use, HCT/Ps product and radiation devices.
Guidance for UDI Implementation
Reporting Databases: Singapore Medical Device Register (SMDR) - For risk class B or higher, Class A Medical Device Database - Risk class A only
Singapore is now requiring compliance with UDI labeling and database registration. They will accept UDI labels for devices already marketed in the U.S. and the EU without any need for modification. However, if they are not marketed in either country, then they are required to implement via Singapore UDI regulations.
Companies are given an additional 6 months from the compliance date to deplete the respective medical devices that have been imported prior to the compliance date and exist in their current supply chain.
Note:
• UDIs will not be required for medical devices for clinical research, investigational testing or clinical trial and custom-made medical devices
• Medical devices authorized for supply via Special Access Routes (GN26, GN27, GN29) are required to comply with UDI requirement on a risk-calibrated approach
Act on In Vitro Diagnostic Medical Devices
Act on Medical Devices
Reporting Database: South Korean Integrated Medical Device Information System (IMDIS)
South Korea has already implemented UDI regulations by Article 20-23 of the Medical Device Act (No. 14330) and Article 54-2 of Enforcement Regulations of Medical Device Act (No. 1512).
Guidance document from Taiwan FDA
Reporting Database: TUDID
Taiwan has previously implemented UDI regulations, which include labeling and database reporting requirements.
FDA website for UDI
Reporting database: GUDID database
The United States has previously implemented UDI regulations, which includes labeling and database reporting requirements.
Each country has their own UDI database and varying requirements for the data stored in those databases. There is overlap in the data required among the various UDI databases, but each country also has unique data they require.
In addition, countries require that UDI-DI information be provided by “issuing entities.” Note that with the exception of China, all countries accept GS1, HIBCC, and ICCBA as issuing entities.
* Data attributes are approximations based on country UDI requirements and include mandatory, optional, mandatory if applicable, and country database auto generated elements.
** Expected to be similar to US GUDID requirements.
Keeping pace with UDI regulations
Keeping track of country-specific UDI requirements, implementation timelines, and affected devices can be a big challenge to RA teams—especially because the information is scattered across many sources and simply hard to find. In this guide, we have consolidated timeline information and device class requirements across multiple countries. While we make every effort to provide accurate and up to date information, it's always advised to check the government website for the country in question.
Additional UDI resources
Looking for more information? You can visit our EUDAMED resource center, where you will find videos and resources to help you plan for UDI requirements in Europe. In addition, you may enjoy our blog post that outlines our views on the recent EUDAMED timeline updates.
For a broader introduction to UDI, see our Rimsys UDI Overview blog post.
If you're looking for an automated, integrated solution to help you meet changing regulations and manage your global UDI program, request a custom Rimsys demo!
%2520(1755%2520x%2520550%2520px)%2520(855%2520x%2520268%2520px).avif)
Why a RIM System is Critical to Successfully Support MedTech M&A Activities
There was significant M&A activity in the MedTech sector in 2024, and the industry is predicting another big year for mergers and acquisitions. As MedTech companies aim to expand their product lines, enter new markets, innovate faster, and remain competitive in a rapidly evolving space, mergers and acquisitions can be attractive and cost-efficient options. Additionally, some manufacturers are choosing to divest parts of their business to hone their focus, drive additional investment in other key areas, and optimize operations.
For the regulatory affairs professional, M&A activities can be anything but efficient. An influx of new products, registrations, and regulatory information to maintain can wreak havoc on RA teams who are already struggling to effectively manage and maintain compliance amid a seemingly constant state of regulatory change.
In an increasingly complex regulatory landscape, many MedTech teams are turning to RIM systems to help them centralize regulatory information across the business, automate time-consuming, manual processes, and strengthen global compliance. These benefits are even more palpable for companies undergoing mergers, acquisitions, and divestitures, giving them streamlined, fully visible regulatory information management that can scale with their evolving business needs and additional regulatory information to manage.
Case Study: Large, Publicly Traded Device Manufacturer Navigates Product Line Divestiture with Ease
A large, publicly traded manufacturer of products for pain management, digestive health, and IV therapy was in the process of divesting one of its product lines to another company. As a result, some of their regulatory employees were transitioned to the company that purchased its product line. The customer leveraged Rimsys’ unique Linked Accounts feature, which allows users to grant external stakeholders controlled access to Rimsys, to give those impacted access to the 100+ registrations associated with the divested products. As a result, the transitioned employees lost no access to their respective products and are able to manage, review, and approve those registrations as they normally would.
“Linked Accounts is a fantastic feature that I didn’t originally appreciate as much. Rimsys made it easy for us to identify the products impacted by the divestiture and provide access to those who need them. It has been a bright spot in the sea of headaches both teams are experiencing when trying to review and approve information in other systems.”
-Program Manager, Regulatory Information Management
When the transaction is complete, the customer will easily be able to export the list of registrations by product tag and archive the registrations in Rimsys for easy management and visibility. The transitioned employees will also still retain access to the information they need in Rimsys as they work to implement their own Rimsys solution to manage those registrations.
Navigating Business and Regulatory Changes with RIM Systems
As the MedTech industry prepares for additional mergers, acquisitions, and divestitures this year, getting a solid regulatory information foundation in place is critical for a successful transition and ongoing compliance. Yet, many MedTech RA teams are using manual processes and siloed systems to manage regulatory information.
One of Rimsys’ goals is to serve as a strategic partner to MedTech RA teams, helping them better understand their current RIM state and the steps they need to advance their processes. This includes the implementation of a RIM system such as Rimsys to centralize their regulatory information, enable easy collaboration with internal and external stakeholders, and automate time-consuming manual processes for strengthened global compliance.
See our Guide to MedTech RIM Maturity, which provides our RIM Maturity Model Framework, for ways to better assess your organization’s current RIM state and incrementally reach new milestones.
Rimsys has helped global MedTech leaders navigate business and regulatory change with unified RIM software that provides full visibility into their regulatory activities. If you’re looking to stay ahead of upcoming strategic activities or are simply looking for a better way to manage your information amid increasing complexity, contact us to learn how Rimsys can help you streamline and automate your processes for long-term success.

Rimsys POV: Updated EUDAMED timeline
The EU Commission has recently announced updates for completing and implementing EUDAMED based on amendment 2024/1860. This article outlines the current EUDAMED timelines and our point of view on these timelines to help industry prepare accordingly.
Current EUDAMED Timelines:
- The target date for the first mandatory application of functional EUDAMED modules is still January 1, 2026. The Vigilance module is expected to be mandatory beginning in Q3 2026 with full EUDAMED functionality planned for Q2 2027.
- The Actor, UDI & Devices, Certificates, and Market Surveillance modules are currently under audit. The independent Minimum Viable Product (MVP) audit is intended to assess and confirm functionality and interconnectivity of the modules that are deemed audit ready. This audit is foreseen to be completed by Q2 2025.
- Mandatory use of each module is to commence six (6) months after the module is declared fully functional through the independent audit and publication in the Official Journal of the European Union (OJEU). The Actor, UDI & Devices, Certificates, and Market Surveillance modules are expected to be declared fully functional at the end of Q2 2025, leading to their mandatory application date of January 1, 2026.
- The Actor, UDI & Devices, Certificates, and Market Surveillance modules are expected to be declared fully functional by the end of Q2 2025 and mandatory for industry use on January 1, 2026.
- The Vigilance module is not part of the ongoing MVP audit and will not be declared fully functional along with the previously mentioned modules. The revised timeline indicates that the audit of that module will occur between Q2 and Q3 of 2025, with the goal of the mandatory application date in Q2 of 2026.
- The development of the Clinical Investigation/Performance Studies (“CI/PS”) module is intended to continue through Q3 2026. An audit to assess the CI/PS module together with the other five (5) modules will be completed once the CI/PS MVP has been developed.
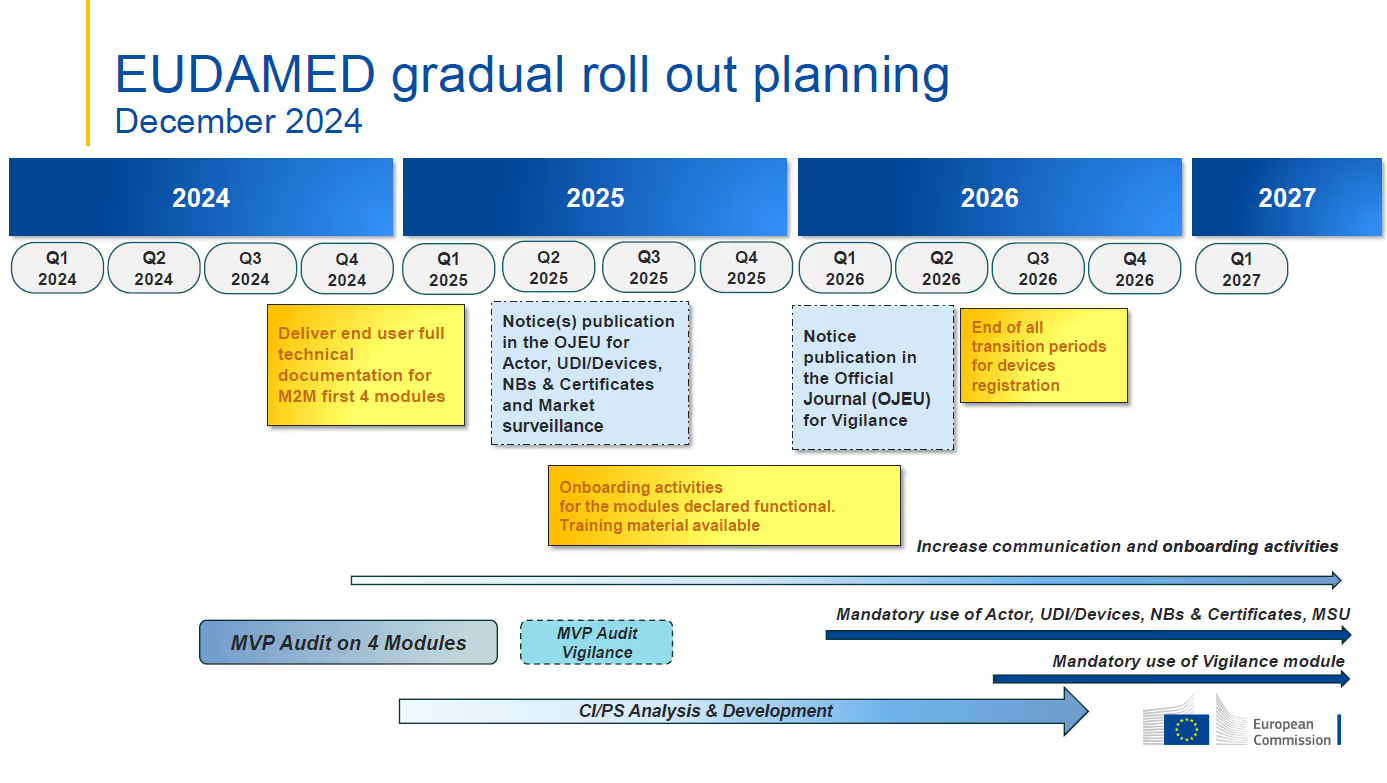
Photo courtesy of the European Commission
Here is how Rimsys views the impact of this announcement for each stakeholder group:
Rimsys
UDI is front of mind as well as future interaction with the Vigilance module. There is no change to our current plans, as Rimsys will continue to develop UDI and Post Market Surveillance functionality regardless of the updated target dates. We also recognize the potential impact of establishing data transfer (DTX) capabilities to interact with EUDAMED in a machine-to-machine (M2M) capacity. With the publication of the final requirements needed for M2M DTX to EUDAMED, Rimsys is positioned to finalize our connection and deliver M2M capabilities as part of the EUDAMED solution.
Industry/customer
Since the European Commission(EC) has made multiple updates to EUDAMED timelines, we expect industry will have some reluctance to accept the new target dates. As a result, this could delay re-engagement with EUDAMED preparations. However, we do not expect the EC to push these updated timelines. Manufacturers that don’t have a plan to submit data to EUDAMED by Q2 this year should expect significant challenges to meet these deadlines. With the audit of expected modules underway with the associated technical documentation published, Rimsys recommends taking steps to organize regulatory data now and submit their information early to all available EUDAMED modules.
EU Commission
The EC strongly recommends industry continues to establish its solution and to submit data on a voluntary basis. Do not wait. The commission’s position is that submitting data early will give companies an advantage by having their data “in” before the onslaught of the entire global MedTech industry, all trying to add data at the same time EUDAMED becomes mandatory. These companies will also be in front of the line to work with the commission resources if data submission issues occur.
* Note - this article includes regulatory interpretations and opinions from the Rimsys team. We try to be as informative as possible, but this information isn’t intended to serve as a substitute for official guidance from regulatory authorities.
