


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.
.avif)

2023 Regulatory performance report
Today at Rimsys, we unveiled the 2023 MedTech Regulatory Performance Report, a new set of insights into the state of medtech regulatory affairs. Compiled based on interviews with 200 regulatory professionals and executives, the study provides a detailed look into how regulatory teams are staffed, their processes, the tools they use, and ultimately how they perform.
Why did we create this study? There were two driving factors behind the research. The first was a common theme that we heard from a number of our customers: Regulatory leaders don’t have clear data and benchmarks. They don’t necessarily know how long a new market submission should take, and how to plan for or assess the work of their teams. While other studies look at the medtech industry broadly or the state of the regulatory profession, this study tries to build a comprehensive resource for regulatory (and company) leaders.
The second factor was really for ourselves and the team at Rimsys. As a company building solutions specifically for medtech regulatory affairs, we wanted more insight into where companies were successful, where they struggled, and where we can add value.
What did we find? Regulatory teams perform a lot of hero work and rate themselves highly for their accomplishments. At the same time there is a lot of opportunity for process improvements, and companies that invest in digital transformation for regulatory affairs see better performance.
Regulatory professionals are superheroes
Regulatory teams are generally pretty small. Most companies have less than 10 full-time regulatory professionals. These small teams complete an enormous amount of work. Last year on average, RA teams completed 50 license renewals, 50 license updates, and 10 new market submissions. This is impressive output.
Digging a bit under the covers, we found that this output relied heavily on the support of external consultants. 90% of companies use consultants to keep pace with their regulatory workload. Front-line employees also struggle with burnout. They were much more likely to report feeling under-resourced than regulatory leaders.
But process problems persist
A lot of regulatory work remains extremely manual. 70% of regulatory teams spend half their time or more on repetitive administrative tasks. All of this manual work increases the frequency of errors and required rework. 61% of companies reported a major non-compliance incident in the past 2 years.
Manual work also makes it difficult to complete regulatory projects in a timely fashion. Teams completed a lot of projects, but each took a long time. Over half of all companies spend 4 months or more on license renewals, license updates, and new market submissions.
Moving regulatory affairs forward
As regulatory requirements become more complex, there’s a natural question about how teams will work moving forward. MDR & IVDR in Europe have significantly increased the regulatory workload required to bring and keep products on the market. Will organizations be able to keep pace with the same resources, tools, and processes?
No, and the performance report shows that medtech companies are investing to improve their regulatory capabilities. The majority of companies are planning to increase the sizes of their RA teams in 2023, and 40% expect to increase their investments in regulatory software. Companies are increasingly adopting specialized software to better support regulatory processes.
Dig into the survey results
The full survey results provide insights into more aspects of regulatory performance. They show that companies need to take a deeper look into their processes and how regulatory resources are allocated. There are two ways to learn more:
- Visit the survey page to see the full results (the survey whitepaper can be downloaded at no cost)
- Watch the recording of our webinar with PA Consulting. We discuss the survey results in more detail and share our regulatory predictions for 2023



Post-market surveillance for medical devices in the European Union
This article is an excerpt from Post-market surveillance for medical device in the European Union.
Table of Contents
- What is post-market surveillance?
- What classes of medical devices require post-market surveillance?
- Components of a successful post-market surveillance plan
- PMS data requirements
- Post-market surveillance system goals
- Required post-market surveillance reporting
- Embracing post-market surveillance as an integral part of your quality program
- Getting started with post-market surveillance
Post-market surveillance (PMS) is designed to monitor the performance of a marketed medical device by collecting and analyzing field use data. Article 10 of the EU MDR and IVDR requires all device manufacturers to have a post-market surveillance system in place. The main elements of the PMS are laid out in Article 83, and additional details for lower-risk and higher-risk devices are covered in articles 84 and85, respectively.
In general, a PMS system consists of both proactive activities and reactive, or vigilance, activities. While post-market surveillance and vigilance are sometimes used interchangeably, vigilance consists of separate activities that feed post-market surveillance programs.
Post-market surveillance systems are used to collect and analyze data not only about the manufacturer’s device but also about related competitors’ devices that are on the market. Data collected through PMS procedures is then used to identify trends that may lead to, among other things, quality improvements, updates to user training and instructions for use, and identification of manufacturing issues.
Note that “market surveillance” encompasses activities performed by a Competent Authority to verify MDR compliance, and should not be confused with the topic of this ebook,“post-market surveillance," which is performed by the manufacturer.
All medical devices marketed in the EU require some level of post-market surveillance, and all medical device manufacturers must implement a post-market surveillance system (PMS). The requirements of the PMS, however, vary and should be “proportionate to the risk class and appropriate for the type of device” (MDR Chapter VII). In particular, the type and frequency of reporting vary based on a device’s risk class.
A post-market surveillance plan (PMS) is an integral part of a manufacturer’s quality management system and provides a system for compiling and analyzing data that is relevant to product quality, performance, and safety throughout the entire lifetime of a device. The PMS should also provide methods for determining the need for and implementing any preventative and corrective actions. A PMS system should include and define:
Surveillance data sources
With the increased focus on proactive risk identification in the MDR, it is important to design post-market surveillance systems that actively acquire knowledge and detect potential risks. It is not sufficient to rely solely on spontaneous reporting by healthcare providers, patients, and other stakeholders.
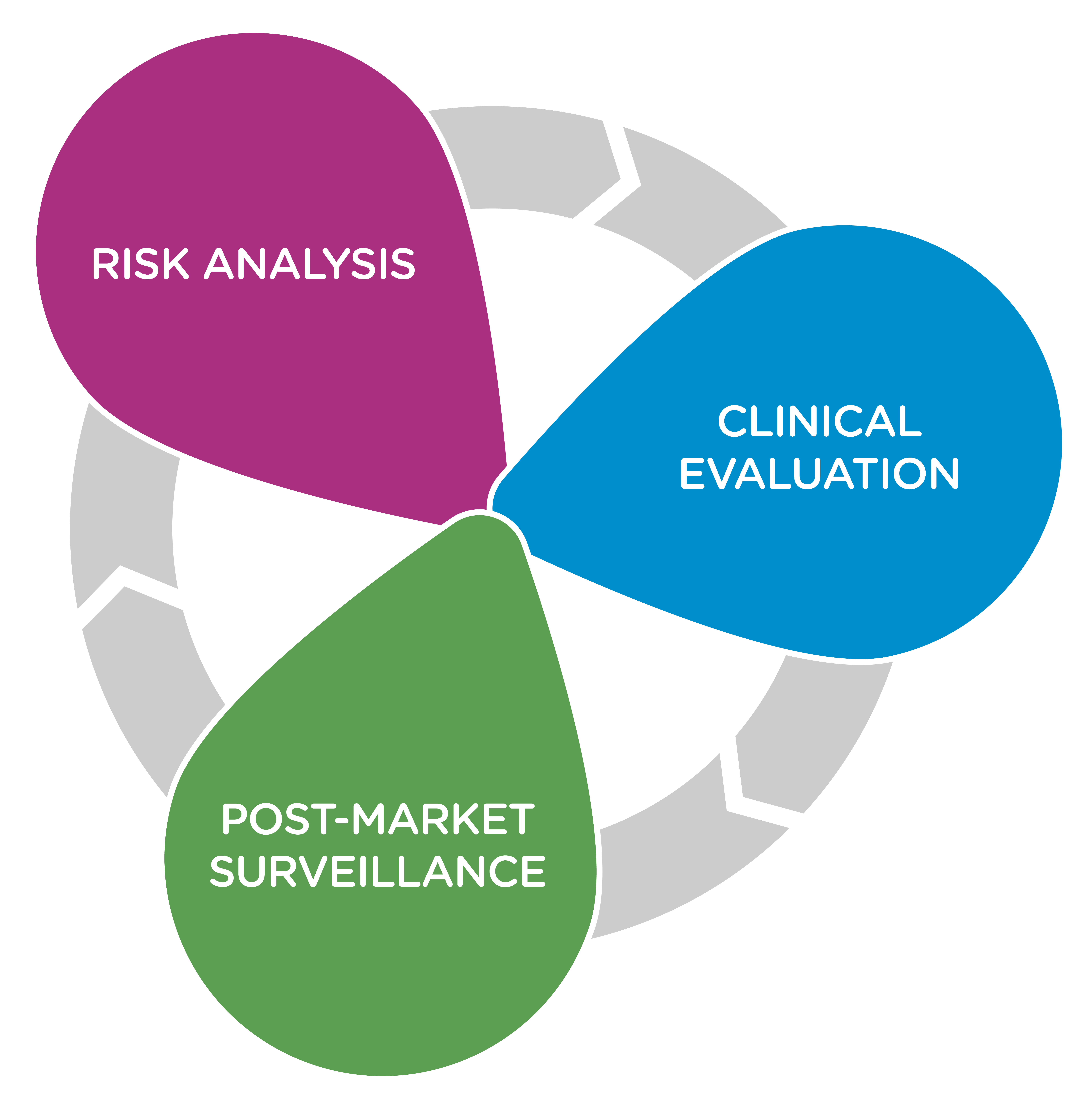
In addition to information coming from Clinical Evaluation Reports and complaint and adverse event reporting, typical sources of surveillance data include:
• Social media networks: Because many of your stakeholders may be communicating on social media networks, it is important to employ social listening techniques and/or tools to identify issues and concerning trends as they develop.
• Industry and academic literature: Any studies, academic papers, and other literature that addresses similar devices or the specific use cases for which your device is designed should be evaluated. In particular, risk factors and adverse events identified with similar devices should be closely examined. It is also important to identify newer technologies that may affect the benefit-risk ratio and establish a new definition of “state of the art” for the device type.
• EUDAMED: While the European Database on Medical Devices (EUDAMED) is not yet fully functional, it is intended to provide a living picture of the lifecycle of all medical devices marketed in the EU. Manufacturers should take special care to consider information for similar devices made available through the EUDAMED system in the future.
• Registries: Patient, disease, and device registries can provide information that informs the clinical evaluation process which provides input into the post-market surveillance system.
Data analysis methodology
A well-defined data analysis methodology will accurately identify trends and lead to defendable decisions in the application of post-market experience. Once the necessary information has been identified and collected, and potentially cleaned of incomplete or otherwise unusable data, the data needs to be analyzed.
The goal is to identify meaningful trends, correlations, variations, and patterns that can lead to improvements in the safety and efficacy of the device. There are many data analysis tools available that can assist with:
• Regression analysis that will identify correlations between data (e.g. the device location/geography correlates to battery life).
• Data visualization that can be useful in spotting trends in the data.
• Predictive analytics, which can be particularly useful with large data sets, to identify future trends based on historical data.
• Data mining, which is also normally used with large datasets, to organize data and identify data groups for further analysis.
Benefit-risk indicators and thresholds
The MDR requires that medical device manufacturers not only demonstrate the clinical benefit of their device but also quantify the benefit-risk ratio. The benefit of a device must be shown to clearly outweigh the risk for it to gain market approval. Article 2 (24) of the MDR defines the benefit-risk determination as “the analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose when used in accordance with the intended purpose given by the manufacturer.”
A PMS system should clearly define benefit-risk calculations and the data used to support them. Post-market surveillance activities are critical in order to re-evaluate and maintain the benefit-risk calculations and determinations of a device throughout its life. Information that is gained through a PMS system can lead to:
• Identification of new risk factors.
• Adjustments to risk frequency and/or severity values based on actual use data.
• Adjustments to established risk calculations based on new “state of the art” technologies becoming available.
• Adjustments to established benefit calculations based on actual use data.
While complaint handling and other feedback tracking are more often described as part of post-market vigilance systems, they play a role in the more proactive post-market surveillance processes as well. A PMS system should define ...
To continue reading this ebook, download the full version.
.avif)
Introducing Rimsys Intel: A Free, Centralized Global Regulatory Intelligence Hub for Medtech
Rimsys is excited to announce the beta launch of its community-driven, centralized hub for regulatory intelligence data, Rimsys Intel. Rimsys Intel builds upon Rimsys’ mission of increasing the availability of life-changing medical technologies by giving users free access to regulatory intelligence, including regulatory affiliations, legislation, UDI requirements, risk class information for medical devices and IVDs, in addition to market access requirements for each regulated country.
As part of our core company value to empower each other, Rimsys believes that regulatory intelligence should be easily accessible and free. I’m thrilled to provide a solution that enables medtech teams to make more informed decisions about market access for their products and execute faster.
To help keep Rimsys Intel up to date amid evolving global regulations, Rimsys is engaging RAPS Regulatory Affairs Certificate (RAC) holders. RAPS, the largest organization of regulatory affairs professionals in the life sciences industry, offers this credential to regulatory affairs professionals who demonstrate proficiency in the scope and application of medical device and pharmaceutical regulations. RAC holders who sign up for and review Rimsys Intel data will have the opportunity to earn recertification credits that count toward maintaining their RAC status.
As Rimsys participates in RAPS Euro Convergence this week, I’m proud to be among its community of inspiring, helpful, and knowledgeable innovators. Our collaboration with RAC holders is a very exciting and mutually beneficial one. Not only is the medtech community able to leverage regulatory intelligence verified by highly regarded RAC holders, but we’re also giving RAC holders a free way to earn recertification credits and further their professional development.
From solopreneurs to enterprise-level medtech companies, Rimsys Intel is equalizing access to global regulatory intelligence data by making it free for the community. Rimsys Intel is currently open to a limited number of beta users. Those interested in signing up for Rimsys Intel can join the beta waitlist here. Rimsys Intel will become generally available later this year.

Evolving global cybersecurity regulations: Challenges and opportunities for medtech teams
The landscape of medical device regulations continues to undergo significant changes globally. Most recently, there have been some noticeable shifts in how regulators are approaching the cybersecurity of medical devices. Recent updates from leading regulatory bodies, including the U.S. Food and Drug Administration (FDA), the European Union (EU), and the International Medical Device Regulators Forum (IMDRF), signal a united front in the drive to enhance the cybersecurity measures of medical devices.
The essence of these updates is clear: Cybersecurity is considered a fundamental aspect of medical device safety and efficacy. The FDA's proposed guidance adjustments, the EU's stringent requirements under the MDR and IVDR, and IMDRF's global harmonization efforts are reshaping the regulatory requirements for a broad range of device types. These changes underscore the importance of integrating robust cybersecurity protections from the earliest stages of device design to their operational lifespan.
With the ever-increasing incidents of security perimeter and data breaches, this transition while warranted, presents challenges for manufacturers to elevate their cybersecurity practices, to innovate with security in mind, and to navigate a complex global regulatory landscape. Yet, it also opens up opportunities to lead in the development of safer, more secure medical technologies that earn the trust of patients and healthcare providers alike.
FDA Cybersecurity Guidances
In the evolving landscape of medical device regulation, the FDA has proposed pivotal updates to its cybersecurity guidance, aiming to fortify the resilience of medical devices against cyber threats. This move reflects the growing interconnectedness of medical devices and the escalating sophistication of cyber threats targeting the healthcare sector. The FDA's draft guidance, "Select Updates for the Premarket Cybersecurity Guidance: Section 524B of the FD&C Act," introduces an entirely new section dedicated to enhancing device cybersecurity throughout its lifecycle. This update emphasizes the criticality of integrating cybersecurity measures from the design phase through the entire lifespan of the device, encompassing premarket authorization, 510(k) clearances, De Novo requests, and more.
One of the significant highlights from the FDA's proposal is the emphasis on ensuring that devices capable of internet connectivity, whether intentionally or unintentionally, maintain stringent cybersecurity safeguards. This perspective stems from an understanding that the ability to connect to the internet inherently poses potential cybersecurity risks. It also expands best practices for cybersecurity within the medical device sector, building on the earlier adoption of a Secure Product Development Framework (SPDF). This framework aims to minimize vulnerabilities in medical devices by incorporating robust processes throughout the product development lifecycle. The guidance also stresses the importance of transparency, urging manufacturers to provide users with comprehensive cybersecurity controls, potential risks, and technical details through labeling. This approach is intended to empower users to manage cybersecurity risks effectively and respond promptly to any identified issues.
In addition to the FDA updates to cybersecurity guidance within medical device regulations, similar positions have been taken by other global regulatory bodies, recognizing the critical importance of cybersecurity in medical devices. As these frameworks get enacted and updated, the industry is seeing a unified drive toward enhancing the cybersecurity of medical devices, reflecting the global nature of both healthcare and cyber threats.
European Union (EU) Cybersecurity Guidelines
The European Union has continued to be proactive in addressing cybersecurity concerns through the Medical Device Regulation (MDR) and the In Vitro Diagnostic Regulation (IVDR). The MDR, which came into full application in May 2021, and the IVDR, fully applicable from May 2022, incorporate specific requirements for ensuring the cybersecurity of medical devices. These regulations require manufacturers to consider cybersecurity at all stages of a device's lifecycle, from initial conception to decommissioning.
More recently, the EU has introduced updates to the Cyber Resilience Act and drafted a new EU cybersecurity rule to establish a European cybersecurity certification scheme (“ECCS”). The ECCS would introduce a detailed certification process, prohibiting self-assessment even for low-risk products. It mandates vulnerability disclosure for certified products, sets rigorous expectations for regulators and certification bodies, including regular product sampling and peer assessments, and requires a proactive approach to vulnerability management. The ECCS also would allow for the mutual recognition of standards internationally and mandate the consolidation of existing national certification schemes. This comprehensive approach highlights the EU's commitment to enhancing cybersecurity across the board.
IMDRF Cybersecurity Guidelines
The International Medical Device Regulators Forum (IMDRF) has also published guidance aimed at harmonizing cybersecurity practices. The IMDRF's guidelines focus on principles for medical device cybersecurity, which include risk management, post-market surveillance, and information sharing amongst stakeholders. These guidelines serve as a reference point for both regulators and manufacturers, aiming to foster a unified approach to addressing cybersecurity risks.
Impact on Device Manufacturers
Manufacturers must navigate these evolving regulatory landscapes, ensuring their devices comply with each jurisdiction's specific requirements. This means incorporating robust cybersecurity measures from the design phase through the entire product lifecycle. Expectations include the ability to update and patch devices in the field, conduct thorough risk assessments, and maintain transparency about a device's cybersecurity measures. The impact of these changes means that medtech design and commercialization pipelines will need to incorporate cybersecurity as a core component, rather than an afterthought. Manufacturers should anticipate:
- Increased Scrutiny: Regulatory submissions will likely require more detailed cybersecurity information, including evidence of risk assessments and mitigation strategies.
- Lifecycle Management: There will be a need for plans to address cybersecurity throughout a device’s lifecycle, including mechanisms for providing updates and patches.
- Global Harmonization: While regulations may vary in specifics from one region to another, the overarching principles of ensuring device safety and effectiveness through cybersecurity measures are consistent. Manufacturers looking to enter multiple markets will benefit from developing products that meet high cybersecurity standards capable of satisfying various regulatory requirements.
The Path Forward for Medtech Cybersecurity
As medical devices become increasingly interconnected and reliant on digital technologies, the importance of cybersecurity cannot be overstated. The FDA’s, European Union’s, and IMDRF’s updates are part of a broader global movement towards securing medical devices against cyber threats. Manufacturers must stay informed about these regulatory changes, integrating cybersecurity into every stage of their device’s development and lifecycle in order to properly comply with regulatory requirements.
Manufacturers and stakeholders should also closely monitor developments in cybersecurity regulations across all jurisdictions where they operate or plan to market their devices. Engaging with regulatory bodies, participating in industry forums, and adopting best practices in cybersecurity will be key strategies for navigating these evolving landscapes successfully and ensuring the trustworthiness and resilience of medical devices in the digital age.
FDA listed, cleared, approved, granted - what IS the difference?
The terms “listed," "cleared,” “approved,” and “granted” all refer to a finding or status from the FDA that authorizes a medical device to be legally placed on the market (for sale) in the United States. As a result, these terms tend to be used interchangeably, but they definitely don’t mean the same thing. Each references a unique pathway to market that is based on the device’s risk class. This article explains the differences between each term and what level of FDA review they require.
Market pathways depend on device classification
A business that is involved in the production and distribution of medical or in vitro diagnostic devices (intended for distribution and use in the United States) is required to register its establishment annually with FDA, using a process called establishment registration. This process requires putting information into an FDA database on their website. This also requires the business to list the devices and the activities performed on those devices, at their establishment. But before you can do this, you need to identify the proper classification of the device(s).
The FDA uses three levels of classifications for medical devices - each carrying a different patient risk value. Once the correct classification is determined, you must then choose the proper registration pathway – Premarket Notification (otherwise known as 510(k)), Pre-Market Approval (PMA), or De Novo process. Before you can legally market your device in the US, it must be FDA Cleared or Approved or in the case of the De Novo process, Granted.
What do the different FDA terms mean?
Regulatory professionals hear the terms Registered, Cleared, Approved, and Granted throughout the medical device industry, and even they are sometimes confused about the differences between them. However, the distinctions are significant, and it’s important to understand those differences and how and when to use them.
- Registered/Listed: A company that has registered with the FDA and has listed their device and the activities performed on those devices at that establishment, into the FDA's registration and listing database. It applies to all class devices, but for most of the Class I devices, it is the only form of registration with FDA. Important to know: the FDA does not issue any type of device registration certificates to medical device facilities.
- Cleared/Clearance: Most of the Class II and some Class I devices require a Pre-Market Notification (510(k)) submission. Before you can sell a device to the public, each submitter must receive an order, in the form of a letter, from FDA which finds the device to be substantially equivalent (SE) and states that the device can be sold in the U.S. This order clears the device for commercial distribution.
- Approved/Approval: A premarket approval (PMA) is the hardest type of device marketing application required by FDA for class III medical devices. To be legally sold on the market, they must undergo an extensive review and approval process. Following a successful submission of a (PMA) or a Humanitarian Device Exemption (HDE), the device is given Approval by FDA.
- Granted: Medical devices using the De Novo process will be Granted approval by FDA before they can be legally marketed in the United States.
Most Class I and some Class II medical devices are exempt from 510k submission requirements.
All other Class II devices require 510(K) clearance as a premarket submission to FDA to demonstrate that the device is safe and effective. Clearance is based on the device being substantially equivalent to an existing, legally marketed device, that does not require premarket approval (PMA). Medical devices in the 510(k) category receive an FDA clearance to bring the device to market.
All Class III devices require a Pre-Market Approval (PMA) - the most stringent type of device marketing application required by FDA. Premarket approval is the required process of scientific review to ensure the safety and effectiveness of Class III devices. Medical devices in this category receive FDA approval to bring the device to market.
Novel devices that don’t have a predicate on the market are classified as Class III by default. However, companies can use the De Novo process to request that the FDA review the risk and safety information of the device for possible re-classification. When a De Novo request is granted, the device is re-classified as Class II, and the device may be brought to market.
Companies can also submit a Humanitarian Device Exemption (HDE) application for Class III devices. A Humanitarian Use Device (HUD) is a device that is intended to benefit patients by treating or diagnosing a disease or condition that affects fewer than 4,000 individuals in the United States per year. The HDE application is like a PMA application, but it is exempt from the effectiveness requirements of a typical PMA.
A relatively newer term being used now is the Emergency Use Authorization (EUA). This is when the Secretary of Health and Human Services declares that there may be circumstances justifying the authorization of emergency use of medical devices, such as during the COVID-19 pandemic. The FDA may issue an EUA to authorize unapproved medical products (or unapproved uses of approved medical products) so that they can be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or conditions when certain criteria are met.
Checking the status of a device with the FDA
The FDA provides several ways to check if devices are approved, cleared, or granted.
To search for FDA-approved or FDA-cleared products by device name or company name:
- Go to the Devices@FDA Database.
To search for FDA-granted products by device name or company name:
- Go to the Device Classification Under Section 513(f)(2)(De Novo) database.
To search for FDA Emergency Use Authorization devices, go to the listing here.
Conclusion
Terminology is only one of the things that can be confusing about the FDA’s processes. Using the wrong terminology can impact your company’s reputation, and possibly have some legal implications, but more importantly, it can mean that you don’t have a clear understanding of how to bring your product to market.
Making sense of the different FDA processes can be challenging—especially for companies that are bringing devices to the market for the first time. For a detailed walkthrough of the steps, documents, and timeline associated with each path to market, see our Beginners Guide to the 510(k), Beginner’s Guide to the FDA PMA Submission Process, and Beginner’s Guide to the FDA De Novo Process.

Learn why UDI is relevant to regulatory affairs and how Rimsys can help medtech RA teams manage the growing complexities associated with UDI data.
This article was last updated March 12, 2024.
What is UDI?
Unique device identifiers (UDI) are now a requirement for medical devices marketed in the US, and are being phased in by the EU and other countries. UDI systems are intended to benefit healthcare providers, manufacturers, authorized health authorities, hospitals and institutions, and individual consumers by providing:
- Faster discovery of possible flawed medical device information by health authorities.
- Quicker access to recall information, and visibility into current inventory.
- A reduction in medical errors through consistently documented product expiration dates.
- Identification of any counterfeit products being used in healthcare facilities.
- Assurances that information regarding an implanted device is safely retained and traceable.
UDI timeframes and deadlines vary by market and product, and have been revised multiple times in some countries. This article details the UDI deadlines for the countries which have announced specific programs, and is current as of the date of this article. Note that these dates can change as participating countries adjust their plans. We will continue to update this as more information becomes available.
Quick Links to country-specific sections:
- Australia UDI
- Brazil UDI
- Canada UDI
- China UDI
- European Union UDI
- India UDI
- Japan UDI
- Saudi Arabia UDI
- Singapore UDI
- South Korea UDI
- Taiwan UDI
- US UDI requirements
- UDI databases by country
The Australian Therapeutic Goods Administration (TGA) announced that mandatory compliance will be progressively phased by device classification, starting with high-risk and implantable medical devices, followed by lower risk class devices over subsequent years. Mandatory compliance will likely not go into effect until the Medical Device Regulations is updated in 2024.
Sponsors and manufacturers can choose to voluntarily comply with the UDI requirements from the date the UDI regulations take effect. Mandatory compliance will commence at a minimum of 12 months from the date the regulations take effect. The reporting database for UDI (AusUDID) is also still in the production phase.
On January 10, 2022, RDC 591/2021, the regulation that requires UDI labeling and database registration for devices regulated by the Brazilian Health Regulatory Agency ANVISA, came into effect. The regulation calls for rolling implementation based on risk class and the establishment of a Brazil UDI database. In June 2024, an amendment to the regulation was published in RDC 884/2024. The updated timelines are published for each risk classes II, III, and IV below. The amendment had no impact on the timeline for class I devices.
In the case of reusable devices for which the UDI information is placed directly on the product, an additional two years have been added to the transition periods below. Details of the UDI reporting database, and related compliance dates, are not yet available. Additional information can be found here: ANVISA UDI guidelines
Health Canada has proposed a UDI framework based closely on the international UDI guidance from the IMDRF. The current proposal involves requiring UDI labeling for all devices, with the exception of Class I low-risk devices. Health Canada intends to either develop a UDI database or modify the existing Medical Devices Active License Listing database (MDALL) to accommodate UDI data.
In addition to labeling requirements, China requires that the UDI be recorded in the China National UDI Database as part of the medical device registration. Additional information on China UDI requirements (link in Chinese) from the China State Drug Agency and Rimsys Ultimate Guide to the China NMPA UDI System.
According to the initial provisions of the European MDR and IVDR regulation, industry use of EUDAMED may not be mandated until all modules are declared fully functional. In the last several months, the MDR/IVDR amendment proposal (23/01/2024) was released to suggest a gradual implementation of individual EUDAMED modules once each has been audited and declared functional. This proposal has been issued with a goal to speed up launch of the modules of EUDAMED as each is finalized to allow for industry implementation and adoption without additional, undue delay. The UDI module of EUDAMED is available for voluntary use currently and, with the provisions of the proposed amendment, could be mandatory use for industry in late-2025 with an expected transition period beginning at the time the UDI module is ready. Additional information on EU UDI system and requirements: EU UDI system and requirements.
At the end of 2021, the Indian Ministry of Health and Family Welfare delayed the implementation of UDI requirements in India and no new deadline has yet been put in place. Originally, Rule 46 of Medical Device Rule 2017 was set to require UDI labeling by January 1, 2022 for medical devices approved for manufacture, sale, distribution, or import in India. Details on how the UDI needs to be displayed and the specific information that needs to be included have not yet been released.
Japan was an early promoter of standardized barcodes, but is still working towards harmonizing their requirements with global UDI expectations.
As of Dec 2022, according to the type of device, bar code labeling based on the international standards is required for immediate containers/wrappings/retail packages of medical devices. It is expected that barcodes would be displayed on every pharmaceutical and medical device in unit of use for patients. Also, safety measures using bar code labeling at clinical settings shall be promoted, as well as registration of production information in the database by MAHs.
Saudi Arabia has allowed voluntary UDI registration since October 1, 2020, but mandatory compliance for class B, C and D devices went into effect September 1, 2023. These requirements apply to both labeling and database (SaudiDI) registration.
Medical devices imported before the compliance date may be distributed without UDI information until one year after the date of full enforceability. This exception does not apply, however, to the Direct Marking (DM) requirement, which is a permanent marking of the UDI on the device itself. For additional information, refer to the Saudi Arabia guidance document.
Singapore is requiring compliance with UDI labeling or database registration regulations based upon classification and a phased in approach. Singapore will accept UDI labels for devices already marketed in the U.S. and the EU, otherwise the UDI will need to comply with all of Singapore’s HSA guidelines, including partnering with an HSA-designated UDI issuing entity. Singapore is also allowing companies a 6-month grace period for medical devices imported before the November deadlines listed below.
Guidance on Medical Device UDI system (GN-32-R2)
UDI compliance is mandatory and was implemented by Article 20 of Medical Device Act (No. 14330) and Article 54-2 of Enforcement Regulations of Medical Device Act (No. 1512). Note that South Korean regulations refer to “Integrated Medical Device Information System,” or IMDIS, which is their UDI database and “Medical Device Standard Code,” which is the UDI code itself. As part of the introduction of UDI, South Korea has also mandated that manufacturers provide a device monthly supply history report, required 1 year from the UDI compliance dates.
South Korean regulations: Guidelines for generating UDIs, Medical Device Act No. 14330 and the Regulation on KGMP No 2016-156 (links in Korean).
Taiwan has already implemented UDI regulations, which includes both labeling and database reporting requirements. The UDI reporting database is referred to as Taiwan UDID (TUDID) and has 23 required data elements. If medical materials meet one of the following conditions, however, then they could be exempt from UDI: Customized medical devices, special medical equipment for export and non-implantable medical device components in the medical device package and in vitro diagnostic medical device package for single use only and not used separately and sold. Read more in the Guidance document from Taiwan FDA.
The United States mandates compliance with both labeling and database requirements for all devices. The FDA does not intend to enforce the GUDID submission requirements for Class I and unclassified devices, other than implantable, life-supporting or life-sustaining devices (I/LS/LS), regardless of whether they were consumer health products, before December 8, 2022.
Implantable, life-supporting or life-sustaining devices, including Class I I/LS/LS devices, should also be complying with GUDID submission requirements. The US FDA requires that all UDI information be entered into the US-specific GUDID database. For additional information, see the FDA UDI system and requirements.
Each country has their own UDI database and varying requirements for the data stored in those databases. There is overlap in the data required among the various UDI databases, but each country also has unique data they require.
In addition, countries require that UDI-DI information be provided by “issuing entities.” Note that with the exception of China, all countries accept GS1, HIBCC, and ICCBA as issuing entities.
Note: * Data attributes are approximations based on country UDI requirements and include mandatory, optional, mandatory if applicable, and country database auto generated elements.
** Expected to be similar to US GUDID requirements.
Keeping pace with UDI regulations
Keeping track of country-specific UDI requirements, implementation timelines, and affected devices can be a big challenge to RA teams—especially because the information is scattered across many sources and hard to find. In this guide, we have consolidated timeline information and device class requirements across multiple countries. While we make every effort to provide accurate and up to date information, it's always advised to check the government website for the country in question.
Additional UDI resources
Our team discussed country-specific UDI requirements and strategies that regulatory affairs teams can use to better manage UDI data in an in-depth webinar. For additional information on UDI requirements, you can watch the webinar replay here, or review our Ultimate Guide to the EU MDR/IVDR UDI and the Ultimate Guide to the China NMPA UDI System.

Key steps to help you streamline regulatory process management
Regulatory affairs is constantly changing, and these changes span the entire product lifecycle - from pre-market awareness of global regulations to managing varying and changing market placement and post-market activities. Managing all types of regulatory changes without streamlined processes and methods in place is a daunting task and one that can set regulatory affairs teams up for risks, including project delays, non-compliances, financial impacts, and employee turnover.
With regulatory affairs interactions and activities often spanning various departments, office locations, and external stakeholders, many regulatory affairs teams are left wondering how they can better streamline their process management.
We recapped core methodologies from our recent webinar, Navigating regulatory change: Why streamlined process management is critical for medtech regulatory teams, to help RA teams review their existing processes, identify gaps, and put together a remediation and implementation plan that will enable them to reduce risks and maintain regulatory continuity.
Phase 1 – Assess your team’s current processes
Assigning resources to provide an honest assessment of your regulatory processes can be tough. We've seen the most success by having awareness and involvement from all of your team’s stakeholders, as many of them have a vested interest in your regulatory processes and information. Additionally, we've seen even greater success when those efforts are supported by an executive sponsor who's committed to streamlining these processes and making the changes.
When we talk about assessing current processes, we don’t just mean reading common SOPs and flow charts. We're talking about an honest review of the adequacy of your regulatory inputs. Some questions you can ask to help with your assessment are:
- Do we have everything that we need when we need it?
- Does this process generate valuable deliverables? How hard are we working to generate these outputs?
- Do we have the right tools in place? If not, what tools on the market can help us achieve our goals?
It’s safe to say you can spend less time on processes that you've identified are working well. If you don’t know exactly where to start in a process assessment, it's helpful to think about where you spend most of your time. When focusing on the processes that are taking up most of your time, it’s easier to see where the inefficiencies lie.
Once the opportunities for improvement are identified, it's time to consider all potential risks. Compliance risk is an important thing to consider, but there’s also business risk with slow processes, inefficient processes, or even worse, ineffective processes where RA professionals spend a lot of time arriving at the wrong output or no output at all.
As risk is assessed for these opportunities, you should then consider the effort that it's going to take to resolve each. This measurement doesn't have to be highly specific. The intent for it is to ultimately help you prioritize which regulatory process changes you want to execute with your team first.
Phase 2 – Planning for Improvement
While there is some planning that needs to take place at the time that you kick off the assessment phase to help you understand and establish who's going to assess which processes., this phase is intended to focus on planning for improvement. It starts with prioritizing your inefficient processes from highest to lowest. Gaps should be prioritized and resourced first without trying to “boil the ocean.” This will help your team set itself up for a successful implementation phase next. From our experience, it’s hard to appropriately focus on process improvement when trying to change too many processes at once.
Within the planning phase, you can review the results of the assessments with all of the regulatory stakeholders and any other stakeholders who are involved with the outputs of the processes. The process owners can then take this opportunity to break down silos by understanding the relationship that their processes have with other departments and the effect their outputs have on these departments. Taking the time to understand the impact your processes have on other departments will allow you to be able to make any necessary adjustments to the inputs and outputs of your processes and hone your communication strategies.
From here, you can assign resources and tasks to manage the overall effort with regular check-ins.
Phase 3 – Implementation
Implementation, the easy part as we jokingly say. As you work through the implementation tasks identified in the planning phase and check in with your stakeholders, it's a good idea to test and iterate on your changes to ensure that they continue to make sense. This is also crucial to verify that your changes are advancing the project or the task toward your overall process management goals.
Training is a key element in the implementation phase as well. Any upfront communication that can be provided to the users of the process ahead of the training is going to be beneficial for change adoption and change management. It’s important to not only communicate that process changes are coming but also why they are coming and what benefits the end users of the process and the consumers of the outputs expect to see as a result.
Once those process changes have been implemented, and training is completed, it’s important to measure and monitor the process for the effectiveness of the changes. Some key questions to consider here are:
- Did we gain efficiencies?
- Did we make our lives easier with these process changes?
- What impact have these process changes had on our team, other departments, and our organization?
If you can’t answer these questions positively, it’s important to go back through the assessment planning phase activities to make sure all improvement opportunities and tasks were properly identified and assigned. When you’ve found those changes have made a positive impact, you should communicate those successes with all relevant teams to build a success story within your organization and to encourage additional adoption of these changes.
Our webinar replay, Navigating regulatory change: Why streamlined process management is critical for medtech regulatory teams, has more tips to help you optimize your process management and explains how regulatory tools such as RIM systems can help RA teams automate, track, and manage their processes across global internal and external teams. Download the full replay here.

Rimsys UDI Overview
UDI: More than Just a Barcode and Label
Unique Device Identification (UDI) is a global requirement mandated by regulatory bodies in various countries to facilitate the easy tracking of key medical device information throughout the supply chain. This system ensures traceability from the moment a device is manufactured until it is used in a medical facility or at home.
The UDI system mandates specific labeling requirements, including the placement of a UDI number, a barcode, and essential device information on the medical device's label. This facilitates the straightforward identification and tracking of the device.
The importance of UDI to regulatory affairs teams
The obligations of the device labeler extend beyond just labeling. Manufacturers are also required to submit and update device information in regulatory databases specific to each country where the device is marketed. This part of the regulation underscores the importance of managing UDI data effectively, as it is critical for legally marketing and maintaining medical devices in different markets. As a result, the responsibility for UDI compliance is increasingly recognized as falling within the purview of regulatory affairs departments within manufacturing companies.

The Unique Device Identification (UDI) system, initially introduced by the US FDA, has since been adopted by regulatory authorities worldwide. These authorities are developing their own UDI programs tailored to their countries to deliver similar patient benefits. Each country's UDI program typically mandates specific labeling and device data reporting and maintenance. While there are overlapping elements for the UDI data required, individual countries have set up additional, localized requirements. This creates a layer of digital complexity through unique regulatory database requirements, interfaces for data entry, and the need for machine-to-machine submissions to handle large-scale reporting.
The increasing blend of shared and unique UDI data requirements, along with country-specific regulatory database needs, highlights the importance of developing comprehensive compliance solutions. The drive for digital transformation in this area is fueled by the intensive data demands from both regulators and manufacturers. This transformation aims to simplify the management of expanding requirements and address the growing complexity as more countries adopt UDI programs.
As UDI programs and the necessity for database reporting become mandatory in more countries, manufacturers and labelers must be ready to establish and maintain UDI datasets for both new and existing products in those markets.
How Rimsys can help
Rimsys regulatory management software offers a platform that simplifies the creation, maintenance, and reporting of UDI data. It also provides tools to oversee and manage the entirety of a company's UDI program through a unified solution.
Business outcomes supported by the Rimsys UDI module:
- Remove the risk of data entry error that comes with keeping identical data sets manually in sync - The Rimsys solution allows users to create and manage UDI attribute data from a centralized location and then apply that information to global UDI requirements, where the data requirements overlap multiple markets.
- Reduce the burden of keeping up to date with each country's UDI program - Rimsys monitors global UDI regulatory changes and adds new country requirements directly into the platform as UDI programs are implemented and become required. Rimsys also keeps up with the latest changes to supported UDI programs for the US (FDA) EU (MDR), Saudi Arabia (SFDA), China (NMPA), South Korea (MFDS), and Singapore (HSA) and updates the required fields directly into the platform.
- An open API ecosystem allows "source of truth" data to be integrated into Rimsys as a "post go-live" phase - Ensure data is up to date and locked at the source of truth, yet centralized for application to global UDI requirements in Rimsys. Data that is required but not controlled in a customer's source system can be managed within Rimsys.
- Built-in support of machine to machine (M2M) transmission to GUDID (FDA) with EUDAMED (EU) coming soon - Rimsys alleviates the need to manually upload UDI data into databases when relevant information changes to ensure compliance. Acknowledgments from machine-to-machine interactions are saved directly to Rimsys and associated to each UDI record.
- Leverage the existing product hierarchy in Rimsys to efficiently manage Basic UDI to reduce non-compliance risk for EU MDR - Data requirements for Basic UDI established in Rimsys are included with the M2M process - Coming Soon
- Facilitate impact assessments in Rimsys since UDI information sits alongside product and registration data - Eliminate the need to manually combine disparate data sets.
Ready to see how Rimsys software can help you create and manage the complexities of UDI data? Schedule a custom demo here.
