


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The beginner's guide to the FDA De Novo classification process
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
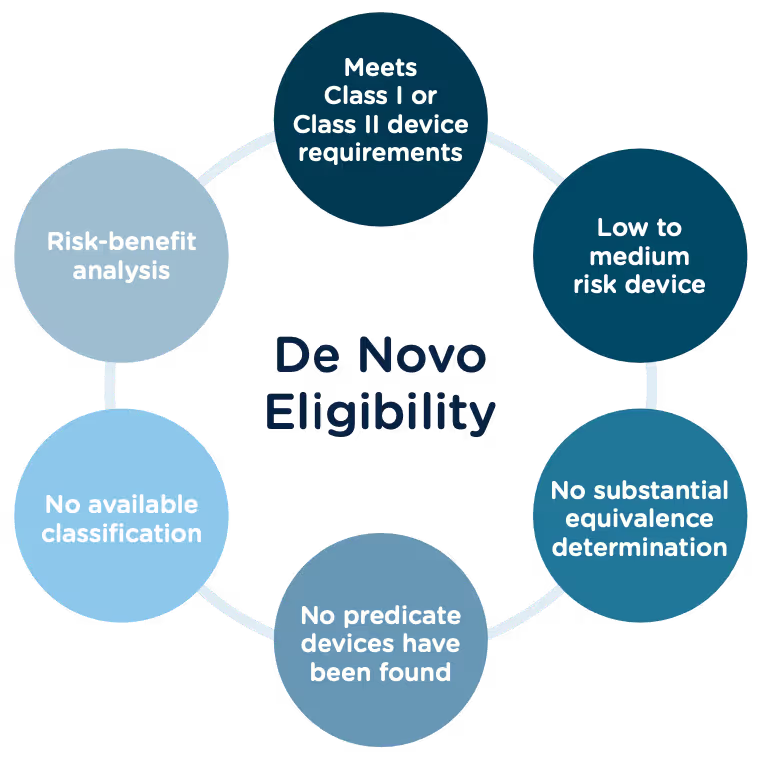
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
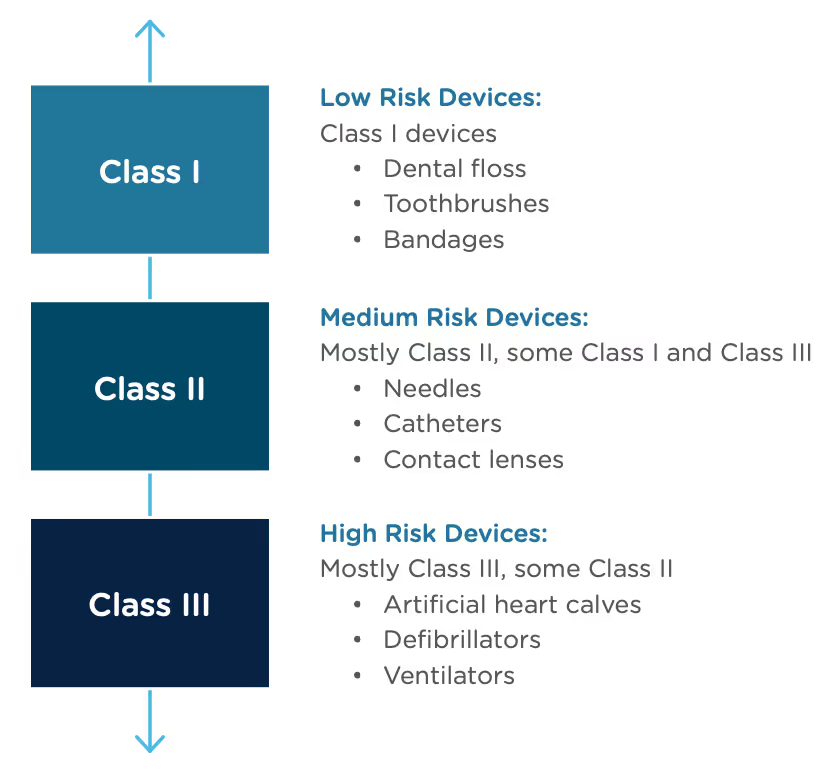
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.



Rimsys secures $1.5 million to accelerate growth of its leading medtech regulatory information management platform
Rimsys Inc., provider of the leading Regulatory Information Management (“RIM”) software designed specifically for medical technology (“medtech”) companies, announced today it closed a $1.5 million investment round, led by Allos Ventures. The financing round will support Rimsys’ penetration of the medtech RIM market through planned expansions of its product offering, sales, and marketing execution. Rimsys serves an expanding list of enterprise customers, including industry leaders such as Johnson & Johnson and Terumo.
"Complex regulatory challenges create barriers that delay product time-to-market, stall revenue growth and increase exposure to compliance risks for even the most experienced medtech companies," said James Gianoutsos, founder & president of Rimsys. "Regulatory teams are in need of digital solutions that provide more efficient and compliant ways to stay on top of a constantly changing regulatory environment"
Rimsys’ solution seamlessly integrates with medtech manufacturers’ existing quality management systems, product lifecycle management systems, and sales and distribution software systems. Rimsys’ robust digital platform, with its intuitive user interface and global intelligence, enables its customers to meet market entrance requirements and grow internationally. John McIlwraith, managing director at Allos Ventures will join Rimsys’ board of directors.
"The maze of global regulations covering medical technology is growing more complex and frequently results in dead ends and delays that can greatly impact commercialization of these products," said McIlwraith. "This financing round will enable the company to further accelerate its momentum with large enterprise customers."
For more information on Rimsys, please visit www.rimsys.io.

Rimsys announces new brand identity for world-leading regulatory information management (RIM) software
Rimsys, a world-leading Regulatory Information Management (RIM) software platform for medical technology companies, announced a new brand identity and tagline. Rimsys, the only holistic RIM software on the market designed specifically for medical technology companies, with functionality for the pillars of regulatory affairs, rebranded to accurately reflect its novel and advanced offering through a distinct, modern brand identity and messaging platform.
The new visual identity, including logo, color palette, graphic elements, and iconography mirror the trusted, relatable, and modern personality of Rimsys. The colors, which feature a vibrant purple and dark blue, were strategically chosen to differentiate Rimsys from other RIM solutions on the market. The visuals are accompanied by a new mission statement, “to digitize, automate, and create regulatory order for the medical technology industry,” and messaging platform that conveys its value propositions:
- Used and trusted by the world’s leading medtech companies
- Built by and for regulatory affairs professionals in medtech
- Easy to start and simple to scale as your company grows
"We’re pleased to announce this rebrand to our customers and partners, with complete confidence that the new identity and messaging accurately reflect our enterprise software solution," said James Gianoutsos, Founder & President at Rimsys.
Rimsys consolidates all the major functions of regulatory affairs on a 100% secure, cloud-based software, making product registration, standards management, essential principles/GSPR, and regulatory intelligence easy. These functions, plus soon-to-be-released features, were formally messaged in the rebrand, including superior server security, system compliance, and simple integrations with many leading enterprise QMS and ERP/PLM software systems.
"With successful implementations in some of the world’s leading medical technology companies including Johnson & Johnson and Terumo, we already know Rimsys has the power to modernize medtech companies of all sizes and scales," said Gianoutsos. "And now, with a brand that mirrors our capabilities, we’re even better poised to reach out and help more."
Components of the rebrand can be seen on rimsys.io, and the full rebrand will be revealed on a new website that is currently under development.

Announcing the release of Rimsys 3.0: Rimsys Insight
Rimsys Regulatory Management Software, the leading and only Regulatory Information Management (RIM) system platform designed specifically for the medtech industry, has just released Rimsys 3.0, which includes its much anticipated regulatory intelligence module, Rimsys Insight.
Rimsys 3.0 now offers curated regulatory news along with changes to laws, regulations, and guidances that are timely and linked to customers’ specific products, markets and the other customizable filters available to each company. Additionally, the integration of market entrance requirements for the top 50+ global markets, further strengthen the already powerful Rimsys Registrations workflow which manages medical device and IVD registrations at the SKU level.
"Information is meaningless unless you can apply context to it as it relates to your company and products. As regulatory professionals ourselves, we know the specific pain that is experienced by our customers. The user experience should be seamless, fully integrated, and intuitive enough that, regardless of how long that person has been in the industry"
Rimsys was built for and by regulatory affairs professionals, enabling teams to digitize and automate otherwise disjointed, paper-based and manual processes. Rimsys has spent the last year working with customers from some of the world’s leading medical device manufacturers to develop the requirements that seamlessly integrate their business processes with its existing ecosystem of cloud-based regulatory affairs software solutions. This includes global product registrations, submissions management, standards management, essential principles and more.
"There’s simply no other holistic and vertically integrated solution quite like Rimsys out in the marketplace. Our initial features were focused on bringing MedTech companies together to collaborate and connect with each other internally. Built on this solid foundation, our latest release truly empowers and connects them to the entire global regulatory ecosystem"
Rimsys is a holistic approach to modernizing, digitizing and automating regulatory affairs activities for the MedTech industry by providing a single source of truth. Rimsys is now aligned and positioned to continue its growth and mission to "digitize, automate and createregulatory order for the medical technology industry™."

5 signs you are ready to take your medical device regulatory team digital
The medical device industry is in dire need of robust, practical and easy-to-use software to make regulatory professionals’ lives easier. Without a unified, collaborative, and regulatory digital system, serious consequences to a business can occur, including an increased risk of noncompliance, increased costs, and a significant reduction in a product’s revenue potential.
Here are the five biggest indicators that it’s time to take your medical device regulatory team digital.
1. You miss simple regulatory compliance requirements
Has your company missed an update to a standard? When a standard is updated that compliance is claimed for your products, a thorough review and associated gap analysis need to be conducted to determine if there is any impact to your products. You also need to go through every single essential requirements checklist to determine where that standard is referenced and update it throughout. If you miss those activities, you will most likely receive a finding when being audited that will require corrective action and significant resources to fix and ensure it doesn’t happen again.
Additionally, as the new Medical Device Single Audit Program (MDSAP) is rolled out, regulators from different markets are now working together to identify instances of noncompliance as well as misalignment of information in submissions and other communications.
MDSAP requires medical device manufacturers to produce evidence of marketing authorization for each country that they sell into. Your regulatory team needs automation to work for them in a way that creates safeguards to prevent unintentional release of products into markets.
There is a high need for more effective control of the submission process, enabled by a unified platform, which can lead to a leaner, higher quality submission and a reduced regulatory burden.
2. Your revenue is impacted
Are you missing registration dates, experiencing slow-to-market losses or long-term, cascading impacts such as loss of customer loyalty? All of these have an immediate and lasting impact on market capitalization.
If your marketing authorization lapses, your company legally cannot continue to sell within that country or market, guaranteeing your sales team becomes frustrated because they won’t hit their sales numbers and your company’s financial projections will be impacted.
Moreover, improper release of a product due to lack of visibility to regulatory statuses can cause fines and loss of credibility with authorities, which can result in increased scrutiny.
Based on a recent survey of 100 companies, 65% of regulatory professionals require at least a week to gather the information needed to determine where their products are sold and whether they are properly registered within a country or market.
If you can’t easily find the information, how effective and compliant can you really be? It may be time to take your regulatory team digital.
3. You miss your time-to-market targets
How do you organize and manage your regulatory information?
If you immediately thought of SharePoint, Excel, Word, Email, Outlook, or Dropbox, you probably aren’t working in the most efficient manner.
In fact, based on a Deloitte study, up to 50% of a regulatory team member’s time is wasted looking for information. Not only is your process inefficient but the way that you manage data and documents from a regulatory perspective is broken.
Based on FDA’s published data, 35% of all 510(k)s submitted to the FDA get stamped with Refuse to Accept (RTA) designation due to simple and avoidable mistakes.
Avoiding these needless delays in getting your products to market should be a top priority to ensure months or years are not added to getting your product the proper clearances.
4. You are burdened by administrative activities
Are you having trouble with efficiency, collaboration, and talent retention on your regulatory team?
Employee turnover on regulatory teams is linked to stress and increases greatly if team members consider processes to be inefficient or wasteful. Being able to perform one’s job efficiently and the perception of being part of a high-performing organization contributes to employee satisfaction and retention.
Regulatory processes touch multiple functional areas of a business in the highly regulated medical device industry. Regulatory teams have been piecing together disparate systems to achieve marginal improvement for years and are notoriously understaffed.
By giving them the right tools, so that they can do their jobs effectively and efficiently, should be priority number one.
5. You rely on institutionalized knowledge
Has a top member of your regulatory team left the organization, leaving you with no idea about what projects they had their hands in, the status of current submissions, broken down communications with external stakeholders, or lost critical tribal knowledge that wasn’t passed down?
Have you ever called that employee at their new company just to “pick-their-brain” because that critical information wasn’t transferred before they left?
Having a fail-safe in place now for when (not if) your top talent leaves prevents the loss of company and product specific tribal knowledge.
Bringing your regulatory processes into the digital age so your team and company can perform work within a central location ensures everything is properly documented and builds that critical archive of information. A unified system and collaboration hub keeps everyone on the same page within a single regulatory platform. Think of it as your company’s regulatory insurance policy.
Employees change roles, leave departments, and move on to other companies, leaving you to pick up the pieces. Retraining a new employee without the subject matter expert can cause delays and wasted time. So stop relying on disparate systems, disjointed processes, color-coded excel files, and emails that get lost in the shuffle and inhibit progress.
The good news
There is good news. Software solutions already exist that create a collaboration hub to help medical device companies actively navigate the changing regulatory landscape.
Software solutions such as Rimsys Regulatory Management Software can empower your current team. They provide better planning and tracking in a unified system that can monitor process metrics, milestones, and automatically inform submissions plan timelines with actual performance. A solution that connects planning to execution, allowing improved, real-time process monitoring provides teams visibility to quickly spot constraints and take action, allowing products to get through your regulatory process faster and with improved compliance.
It is time to set the new standard for the industry. It is time to go digital and start automating your broken systems.

Announcing the Rimsys advisory board
Rimsys Regulatory Management Software, the leading Regulatory Information Management (RIM) platform designed specifically for the medical device industry, is proud to announce the creation of its prestigious advisory board.
By creating an advisory board with the most forward-thinking minds and preeminent talent in the medical device industry, Rimsys is now aligned and positioned to continue its growth and mission as the leading regulatory management software in the medical device industry.
The board members serve as strategic partners in the continued development and success of Rimsys, as a catalyst to achieving its short-term and long-term goals. The board is comprised of trusted thought leaders, known for being change agents in the industry and having the respect of their peers throughout their career and community.
"The management team could not be more pleased with the addition of these board members and involvement they have with the direction of our company. We are fortunate and thrilled to have such talented and experienced industry veterans and look forward to their many contributions,” said James Gianoutsos, Founder & President of Rimsys."
As advocates and ambassadors of Rimsys, the board supports the management team through strategic analysis, consultation, and providing professional expertise and guidance to help navigate and mitigate potential risks, discover opportunities, and define benchmarks for continued success and organizational growth.
"Rimsys is a very unique product in the marketplace, so it’s only fitting that we bring on such unique minds to the board. Their expertise and vision are exactly what is needed to help us improve our business, our technology and expand our product offerings,” said Brad Ryba, CTO of Rimsys."
The current advisory board members include:

John Speer
Jon Speer has over 20 years of experience in the medical device industry that includes quality management, product development, and project management at Creo Quality, Cook Inc., Theron Inc., and Maetrics LLC. Jon is experienced in managing multiple projects and taking medical device concepts through development, regulatory submission, and ultimately to market. Additionally, Jon is an expert in the design and implementation of FDA-compliant quality management systems and is an active contributor at MedCity News, Med Device Online, Quality Digest, QMed, and is the host of the #1 most downloaded podcast in the industry, The Global Medical Device Podcast. Jon currently serves as the Founder and VP QA/RA at Greenlight Guru, an eQMS that is specifically designed for the medical device industry.

Chris Ferguson
Chris Ferguson has over 15 years of global medical device quality and regulatory affairs experience managing class I, II, and III medical devices and consumer products for numerous world-class global organizations. Chris has successfully led global quality and regulatory projects and teams through FDA, ISO, consumer safety audits, and quality system remediation activities and has in-depth knowledge of the current regulatory landscape. Chris currently serves as Director of Quality Assurance for TransEnterix, Inc.

Bruce McKean
Bruce McKean has over 25 years of medical device industry experience as a regulatory professional specializing in quality and regulatory (Q&R) compliance, design controls, and Q&R related mergers and acquisitions. During his career, Bruce has focused on implementing and maintaining design controls, product submissions, quality management systems internal to his company and for newly acquired companies, corporate Q&R internal audit program, and performing Q&R due diligence audits on target companies. Most recently, Bruce has led a corporate-wide MDSAP compliance initiative and is focusing on the EU MDR implementation. Bruce currently serves as Director of Q&R Operations at Philips Healthcare.

Adam Price
Adam Price has over 15 years of medical device industry experience as a quality assurance and regulatory affairs professional. Adam is currently focused on the development of strategies and solutions to establish and maintain compliance in today’s fast-paced regulatory environment to enable businesses to meet the demands of the global market. Adam is cognizant of dynamic and complex market requirements and the need for effective tools and solutions to allow businesses to maintain continued regulatory compliance. Adam currently serves as Head of Post-market Surveillance at Philips Healthcare.

The 510(k) application: if content is king, then communication is queen
Often, the first thing we hear from a consultant or a medical device company regarding an FDA 510(k) premarket notification is that it was delayed because the FDA reviewer did not understand something simple within the application, or completely missed it.
What is wrong with the reviewer? How could they have missed something so simple? I couldn’t have been any clearer!
Sound familiar?
FDA is overworked, under-resourced, and will most likely miss something simple in your file upon reviewing.
As the specification developer, you know the design and history of the product better than anyone. You are providing that entire history in a formal application for review, and hopefully, clearance. A basic understanding of the technology is a must; however, think about the situation from the FDA reviewers’ point of view. 510(k) applications are inherently technical and sometimes need a brief discussion with the FDA reviewer for clarification or a general overview of your device.
Starting this dialog earlier is important for a smooth path to clearance. Part of this process involves requesting a Pre-Submission (“Pre-Sub”). Pre-Subs are a type of feedback that is part of FDA’s Q-Submission program.
Pre-Subs
Pre-Subs are a formal written request from an applicant for feedback from FDA to be provided in the form of a formal written response or a meeting (in-person or teleconference) in which the feedback is documented in meeting minutes. A Pre-Sub provides the opportunity for a submitter to obtain FDA feedback prior to intended submission of a premarket submission (i.e., IDE, PMA, HDE, De Novo request, 510(k), Dual, BLA, IND) or Accessory Classification Request, among others.
Pre-Subs are entirely voluntary on the part of the applicant. However, early interaction with FDA and careful consideration of FDA’s feedback may improve the quality of subsequent submissions, shorten total review times, and facilitate the development process for new devices.
Pre-Subs provide FDA reviewers with an introduction to you and your device rather than just having a 510(k) application dumped on their desk. FDA reviewers appreciate Pre-subs because they can get a sense of when they should anticipate filings and can plan their workloads accordingly.
FDA reviewers, like all of us, only have a certain amount of time during the day. If they are unable to find information easily or do not properly understand something, then they may state that the relevant information is missing from the application or needs further clarification. This kicks the 510(k) application back to you and stops the review clock. That is directly on the industry submitter, not the FDA reviewer.
The bottom line
FDA reviewers are people too. This is an obvious but often overlooked point to make. Sometimes they miss simple (and sometimes seemingly apparent) information. They make mistakes. The last thing you want to do is start yelling or pointing fingers. After all, you don’t want to burn any bridges as you will most likely deal with the same FDA reviewer upon subsequent submissions for similar products. Always be timely, concise, straightforward and respectful.
At the end of the day, keep in mind that your FDA reviewer isn’t as familiar with your medical device as you are. You need to help them understand items that are unclear, and the only way to do that is through building the communication channel early and having constructive conversations.
Did you know Rimsys Regulatory Management Software will keep track of all communications, notes, decisions, and tasks associated with your 510(k) application and other international regulatory submissions? Find out more now with a free demo and we will show you the power of the only regulatory information management (RIM) system platform designed specifically for the medical device industry.
