


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The beginner's guide to the FDA De Novo classification process
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
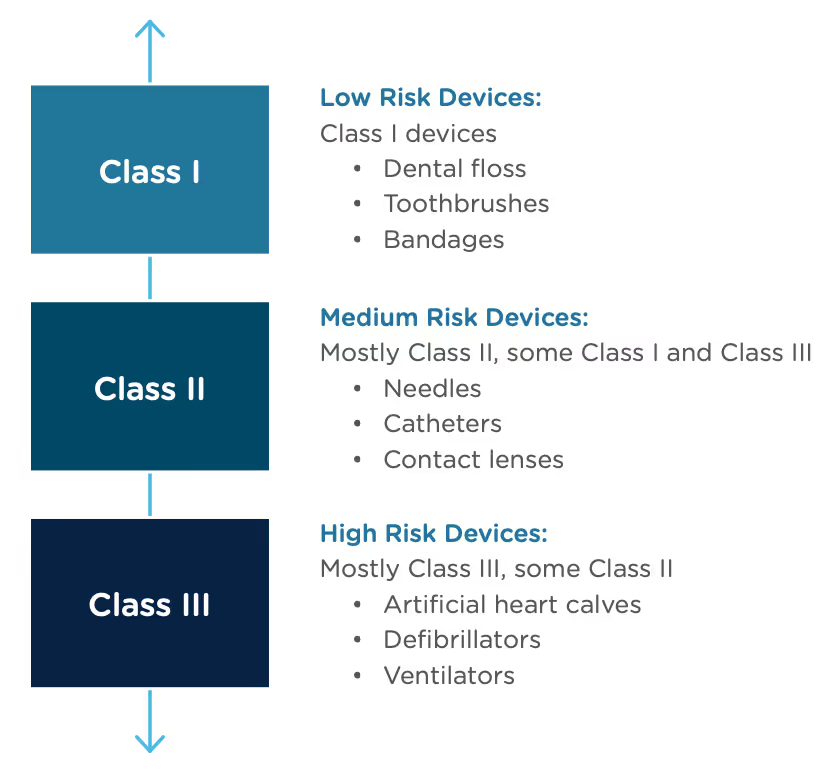
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
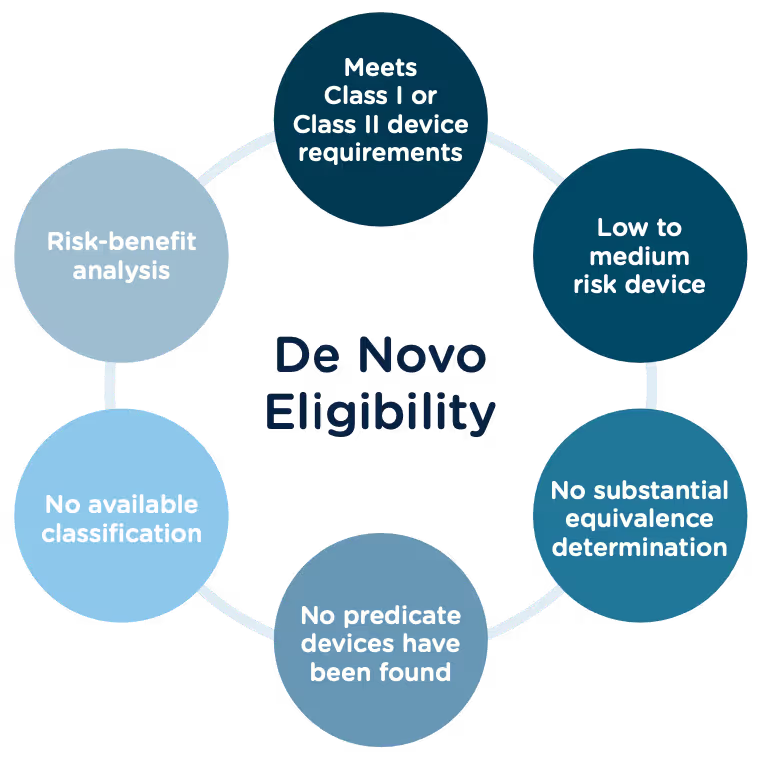
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.




Introducing government submission templates (i.e. 510k, STED, CSDT) and more!
Rimsys released a major revision on Dec. 3, 2018 that included adding registration workflows, registration owners, Kanban boards, new registration dates (e.g. anticipated approvals dates) and registration lifecycle stages.
Rimsys has been working aggressively over the last month to finish up the final touches on our next release, and we are excited to tell you that it has been officially released! These new features will benefit any size of an organization and continues our pathway to better serving the regulatory affairs professionals in the medical device industry.
Here are a few of our features released:
- Document templates – Depending on where you are registering your product, you can now choose or create your own document template that your team can follow to keep you compliant, better organized, and standardize your regulatory process. A few of our templates include: Summary of Technical Documentation (STED) for IVD and non-IVD Medical Devices, ASEAN Common Submission Dossier Template (CSDT), 510k Template, and more!
- Multi-product registrations – You can choose 1 or 1000 products (at the part number level) to register simultaneously into one market.
- Bulk search & replace for essential principles – We have been working with a few of our customers to get this functionality rolled out by the beginning of January. You now have the ability to search / replace / or remove a standard or a document throughout multiple essential principle tables simultaneously. Let’s say you are managing 10 (or even 500) essential requirements checklists…with a few clicks of a button, you can search, find and replace 1 (or all) standards or documents in EVERY table! If you have ever managed an essential requirements checklist before, we can’t stress enough of how HUGE of a time saver this is for you and your team!
- Embedded documents in essential principles – We now automatically embed your objective evidence directly into the Essential Principles PDF record. This means that when you export your essential principles as a PDF, every single document that is linked to it will be embedded directly into the searchable PDF. You never have to go looking for documents again!
- Dashboard updates – Added key metrics so your team can all be on the same page
- Expanded reporting capabilities – Added the ability to drill-down into key metrics
With this release, Rimsys will be better positioned to cater to organizations of all sizes. We have even more features and modules coming out in the coming months that will further enhance the benefit you receive from using Rimsys.
What’s next?
Rimsys has been working hard to be the single source of truth of all things regulatory related for medical devices. One of the most frequently requested features from our customers is the ability to bring regulatory updates on regulations, laws and guidance documents directly into Rimsys. We are happy to report that this feature has been in development for quite some time and we will be releasing in the next couple of months.

MDSAP device marketing authorization and facility registration
What is the medical device single audit program (MDSAP)?
The International Medical Device Regulators Forum (IMDRF) recognized that a global approach to auditing and monitoring the manufacturing of medical devices could improve their safety and oversight on an international scale. This created the Medical Device Single Audit Program (MDSAP) and allows a recognized Auditing Organization to conduct a single regulatory audit of a medical device manufacturer that satisfies the relevant requirements of the regulatory authorities participating in the program.
To date, the MDSAP participating countries include:
- Australia (Therapeutic Goods Administration – TGA)
- Bazil (Agência Nacional de Vigilância Sanitária)
- Canada (Health Canada)
- Japan (Japanese Pharmaceuticals and Medical Devices Agency)
- United States (FDA)
The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Programme and the European Union (EU) are Official Observers, which means they are waiting for the results of the pilot MDSAP program to determine if it’s worth their while to sign on as an official partner.
When does MDSAP come into effect?
Starting January 1, 2019, if you’re selling medical devices into Canada, it’s not optional and you must be certified to MDSAP, or at the very least, show evidence that you are in the process of complying.
As part of the MDSAP auditing program, there are seven chapters an auditor must cover. One of those chapters is specific to marketing authorization and facility registration, which also touches on two other chapters, management and design development. An auditor will be specifically looking for the following:
- Have you complied with requirements to register and/or license your device facility;
- Did you submit device listing information;
- Did you obtain device marketing authorization;
- Have you arranged for assessment of changes and obtained marketing authorization for changes to devices or the quality management system which require an amendment to existing marketing authorization
You must have that information organized in a meaningful way that you can get to it quickly and show, objectively, that you fulfilled the requirements of MDSAP and all of the country regulatory requirements that fall under MDSAP. That also goes hand-in-hand with ISO 13485:2016 where you need a controlled release of products into the appropriate jurisdiction. If you’re trying to be a global leader or a global company, for that matter, in this day and age, you need to have a solid system in place to manage those marketing authorizations worldwide.
Controlled release of product
If you are selling out of the United States, you must comply with the laws of each importing country. That simply means, no matter where you sell outside of the United States, you must meet the importing country’s requirements for marketing authorization. Your regulatory team and business need to be on point by having a robust regulatory system in place that upon product release, you’re meeting those specific requirements. You must have a mechanism in place to ensure that you don’t release product prior to it being properly registered.
That mechanism starts during product realization. Sales, marketing, customer service, engineering, operations, and regulatory teams must all be on the same page. Often times, regulatory is perceived as the bottleneck to product release. However, this is a misconception and is primarily driven by poor planning during the design and development process.
Auditing to MDSAP
Auditors are looking for the standardized process for controlling the release of the product and ensuring that the process has been adequately established and implemented within your facility. MDSAP has a very rigid auditing process to ensure the proper market authorizations have been obtained and facility registrations have occurred.
When your company is audited, an auditor will request records from product outside of the MDSAP participating countries due to the broad jurisdiction of US and international regulations. If the auditor finds issues with those products, they can draw that parallel to determine that your company doesn’t have a controlled product release process and you need to investigate to ensure there isn’t a systemic issue. That means an audit observation and a corrective and preventive action (CAPA) plan need to be established to rectify the issue(s).
What does this mean for medical device manufacturers?
A regulatory professional’s job is worldwide nowadays, which means it is a lot of responsibility, burden and business risk that are on their shoulders. Do you really want all of that being managed by excel files, outlook reminders, and disjointed processes? It must be a fundamental, standardized process, ingrained into your quality management system, that you need in place in order to NOT run into any compliance issues. Your organization must have a standardized process to ensure that your company is releasing good (and approved) product into the market while maintaining any changes to that product (and registration) while it’s in that market.
The requirement is not only that you get the marketing authorization, but you stay compliant when you’re already in that market. That means you must constantly be monitoring for expiring registrations, any type of design changes with your product, and how they affect your marketing authorizations within those countries.
From a quality management system standpoint, you need a good change control process in place that ties directly to your regulatory team. If you don’t have a good regulatory process now, you’re not going to have one later. It’s going to be too late, and the amount of information that your regulatory team must handle today is only going to increase. That’s why you must develop those systems now.
To learn more about the MDSAP, markets where it’s applicable, pros and cons of using MDSAP vs Regulatory Authority inspections, and audit sequence and grading, download our Ultimate Guide to MDSAP.

Top 6 benefits of a regulatory information management (RIM) system for medical devices
The medical device and in-vitro diagnostic medical device industry are in dire need of a robust, practical and easy to use regulatory information management (RIM) system. Without a unified and collaborative system, serious consequences to your business can occur, including an increased risk of non-compliance, increased costs as well as a possible significant reduction in a product’s revenue potential.
1. Revenue impact
Missing registration dates, slow-to-market losses, and long-term, cascading impacts such as loss of customer loyalty have an immediate impact to market capitalization. Moreover, improper release of product due to lack of visibility to regulatory statuses can cause fines and loss of credibility with authorities, which can result in increased scrutiny.
2. Regulatory compliance
Compliant product releases are required in the medical device industry. Automation that creates safeguards to prevent unintentional release of products into markets is a must. Regulators from different markets are working together to identify instances of non-compliance as well as misalignment of information in submissions and other communications. More effective control of the submission, enabled by a unified platform, can lead to a leaner, higher quality submission and a reduced regulatory burden.
3. Faster time to market
Better planning and tracking in a unified system can monitor process metrics, milestones, and automatically informing submissions plan timelines with actual performance. A unified solution connects planning to execution, allowing improved, real-time process monitoring. Teams can quickly spot constraints and take action, allowing the product to get through your process faster.
4. Efficiency and collaboration
Regulatory processes touch multiple functional areas. Regulatory functions have been piecing together disparate systems to achieve marginal improvement. This landscape inhibits the accurate and timely transfer of data and disruption in cross-functional workflow.
5. Efficiency and collaboration
Employee turnover on regulatory teams is linked to the stress and increases greatly if team members consider processes to be inefficient or wasteful. Being able to perform one’s job efficiently and the perception of being part of a high-performing organization contributes to employee satisfaction and retention.
6. Insurance policy
Having a fail-safe in place now for when (not if) your top talent leaves prevent the loss of company and product specific tribal knowledge. Retraining a new employee without the subject matter expert can cause delays and wasted time. A unified system keeps all information within the company.

Introducing project management and essential principle templates
Directly on the heels of our new User Interface (UI) released at the end of September and debuting at the RAPS Regulatory Convergence in October, we are proud to announce another HUGE release.
Project management (for large and small teams)
Properly managing registrations across the world with dozens of stakeholders trying to collaborate on critical information is challenging enough, so we just made it easier for enterprise and large teams to manage everything.
The project management features were frequently requested and will set Rimsys up for further development of Key Performance Indicators (KPIs) to monitor the efficiency of your team and the registration process.
It will also allow Rimsys to further expand reporting capabilities and dashboard metrics so you can easily track and analyze data specific to your team, registrations, products, and countries.
- Registration owner - Assign an owner so you know who is responsible for each registration
- Anticipated approval date - Identify an anticipated approval so you can forecast product releases with other departments
- Registration start date - Automatically creates the registration start date so you can monitor exactly how long a registration takes from start to finish.
- Registration lifecycle stages - Whether you are in the discovery, planning, execution or submission stage of the registration process, you can now keep track with your own configurable buckets.
- Kanban boards - Visually see your registrations in each lifecycle stage and transition them into new stages by a simple drag n’ drop interface.
Essential principles (expansion of templates)
We always had a grander plan in mind. Rimsys was originally set up to include the new EU Medical Device Regulation (MDR) 2017/745 Annex I General Safety and Performance Requirements (GSPR) but now supports Essential Principles Templates that include:
- IVDR 2017/746EU IVDR GSPR
- Australian (TGA) Essential Principles
- Japan (PMDA) Essential Principles
- GHTF/SG1/N68:2012 IMDRF Essential Principles
- Directive 2006/42/EC – Machinery Directive
- and more to come!
The essential principles expansion complies with country entrance requirements and will set Rimsys up for further development of correlation tables. What are correlation tables you ask? Think of this….you create the general safety and performance requirements table for the EU MDR, then with a click of a button, you create the essential principle tables that meet the requirements for all other countries. More to come…
What’s next?
Next month, we will be making a few more major announcements that will bring you new and even better features that will drastically create more value to your company and team. We can’t wait to share the news with you…stay tuned!

Is a medical device accessory a medical device?
Rimsys’ own James Gianoutsos recently contributed an article on www.meddeviceonline.com discussing FDA’s guidance document describing accessories and classification pathways.
On Dec. 20, 2017, the FDA issued Medical Device Accessories – Describing Accessories and Classification Pathways: Guidance for Industry and Food and Drug Administration Staff, which applies to the Center for Devices and Radiological Health (CDRH) and Center for Biologics Evaluation and Research (CBER) for combination products.
The guidance document offers welcomed clarity on the role of an “accessory” and its regulatory relationship to its parent device. As always, guidance documents are not legally enforceable; rather, they describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited.
The guidance explains which devices FDA generally considers “accessories” and describes the processes under Section 513(f)(6) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) to allow requests for risk- and regulatory control-based classification of accessories. In other words, it specifically details what is and is not an accessory, as well as the regulatory routes to classification.
The updated guidance was derived from an August 2017 amendment to section 513(f) of the FD&C Act (FDA Reauthorization Act of 2017 (Pub. L. 115-52)) to state that “the Secretary shall … classify an accessory under [section 513] based on the risks of the accessory when used as intended and the level of regulatory controls necessary to provide a reasonable assurance of safety and effectiveness of the accessory, notwithstanding the classification of any other device with which such accessory is intended to be used.”
The amendment allows for some accessories to have a lower risk profile than that of their parent device and, therefore, may warrant being regulated in a lower class. As classifications for accessories are now risk-based, it provides manufacturers with regulatory flexibility to loosen some of the regulatory burdens on accessories that may not have the same risk profile as their parent devices.
For example, an accessory to a class III parent device may pose lower risk that could be mitigated through general controls, or a combination of general and special controls, and thus could be regulated as class I or class II. A common example of this would be a ventilation system (parent device) with a face mask (accessory).
Additionally, the guidance details the applicable definitions within Section IV: Definitions:
- Accessory — “A finished device that is intended to support, supplement, and/or augment the performance of one or more parent devices.”
- Component (21 CFR 820.3(c)) — “[A]ny raw material, substance, piece, part, software, firmware, labeling, or assembly which is intended to be included as part of the finished, packaged, and labeled device.”
- Finished Device (21 CFR 820.3(l)) — “[A]ny device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized.”
- Parent Device — “A finished device whose performance is supported, supplemented, and/or augmented by one or more accessories.”
Accessory classification policy
The risks of an accessory are the risks it presents when used with the corresponding parent device as intended. To classify an accessory, FDA addresses the following two questions:
- Is the article an accessory? This can be answered by determining the intended use of the accessory. Is it intended for use with one or more parent devices, and does it support, supplement, and/or augment the performance of one or more parent devices?
- What is the risk of the accessory when used as intended with the parent device(s), and what regulatory controls are necessary to provide a reasonable assurance of its safety and effectiveness? This can be answered by providing a detailed risk assessment, outlining the potential hazards and reasonable regulatory and quality controls necessary to assure the accessory’s safety and effectiveness.
Individual accessories may be classified pursuant to the same regulation as a corresponding parent device, when appropriate, or be regulated independently.
Once an accessory has been classified, there is another consideration manufacturers need to decide: the Unique Device Identifier (UDI) rule. Not only does your parent device need to have a UDI, but any and all accessories each need to be assigned a UDI.
As 21 CFR 801 Labeling, Subpart B, Section 801.20(a) states: “(1) The label of every medical device shall bear a unique device identifier (UDI) that meets the requirements of this subpart and part 830 of this chapter,” and “(2) Every device package shall bear a UDI that meets the requirements of this subpart and part 830 of this chapter.”
Further, 21 CFR 830 UDI, Subpart A, Section 830.3 defines “finished device” and “device package” as follows:
- Finished device* means any device or accessory to any device that is suitable for use or capable of functioning.
- Device package means a package that contains a fixed quantity of a particular version or model of a device.
*Note that, although “medical device” and “finished device” are not consistent terminology used within the chapters, the terms are one and the same.
In short, any sellable finished device must bear a UDI, either on the device itself, on the device package, or both. Components to the finished device (i.e., service components and spare parts kits) are not considered accessories, and therefore are not required to bear a UDI.
What does this ultimately mean for manufacturers?
If there was any confusion as to whether a specific accessory is classified as a medical device, it has now been clarified, or at least partially clarified, depending on your specific situation. If there is still confusion among your engineering and regulatory teams, FDA recommends contacting them, via the accessory classification process outlined in the guidance, to classify the accessory appropriately. FDA will treat each accessory classification request as a Q-Submission. Requests may be for a new accessory type (new classification), an existing accessory type (reclassification), or classification of a new accessory type through the de novo process.
A gap analysis should be performed to identify a thorough and complete list of your current and future accessories to determine applicability to the guidance document. Justification also should be documented, should an accessory not apply to the guidance document. Additionally, internal procedures and the process associated with assigning UDIs may need to be updated to ensure there are no compliance gaps.

Podcast – Streamlining the MDSAP device marketing authorization and facility registration process
I had the opportunity to sit down with Jon Speer, Founder & VP of QA/RA at Greenlight Guru to record a podcast to discuss streamlining the MDSAP Marketing Authorization & Facility Registration Process.
If you are not familiar with this topic, you need to hear this.
We discuss:
- Why your regulatory team may be perceived as a bottleneck and why it is important everyone needs to be on the same page about when the product can be released.
- The connection between marketing authorization and facility registration required for various countries – United States, European Union, or elsewhere.
- The need to get organized, get better systems in place, and stay compliant when an auditor comes through your door or when you plan to sell into markets.
- Why small and large companies need to get organized now (i.e. small companies have too much information to maintain, organize, and track while large companies may have resources but suffer from miscommunication and disjointed processes.)
- The need to not be complacent and not be afraid to change systems. Rimsys was created to help regulatory professionals successfully and efficiently handle documentation.
You can hear the podcast below or at the Greenlight Guru blog.
