


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.

The ultimate guide to the EU MDR/IVDR unique device identifier (UDI) System
This article is an excerpt from The ultimate guide to the EU MDR/IVDR UDI ebook.
Table of contents
- Overview
- UDI basics and benefits
- UDI format requirements and issuing entities
- UDI rules for specific device types
- Implementation of UDI and UDAMED in the European Union
- US vs EU UDI comparison
The EU Medical Device Regulation (2017/745) (“MDR”) and EU In Vitro Diagnosis Regulation (2017/746) (“IVDR”) introduce two new systems for information exchange: UDI (Unique Device Identifier) for device identification and EUDAMED (European Databank on Medical Devices) to centralize and disseminate information. UDI is a specific code assigned to all devices and higher levels of packaging. This will allow for devices being sold in the European market to be identified and traced through a globally harmonized approach. EUDAMED is the IT system developed by the European Commission to replace the EUDAMED2 database previously in place under the Medical Device Directives (MDD). EUDAMED is a multi-functional system that will be used to coordinate device registration, provide information about devices to industry professionals and the public, and highlight necessary safety details.
The EU MDR and IVDR UDI system is based upon the guidance of the International Medical Device Regulators Forum (IMDRF). It’s a globally harmonized system that’s designed to increase patient safety and optimize care.
UDI system goals
Increase patient safety
- Improve tracing of devices
- Reduce the presence of counterfeit devices
Ensure access to accurate information
- Unambiguous identification of devices throughout distribution and use
Improve post-market surveillance
- Improve accessibility of adverse event reports
Enhance supply chain Management
- Streamline supply chain process and inventory management
- Simplify medical device documentation processes
The UDI system has four key elements
Element 1: Assignment of UDI (UDI Components)
The first element of the UDI system is the assignment of a UDI. The UDI is a code of alphanumeric characters that acts as the access key to information about a specific medical device on the market. The EU MDR and EU IVDR requires that a UDI be assigned to all medical devices except for custom-made or investigational devices. There are three components of a UDI:
- Basic UDI-DI
- UDI (consisting of UDI-DI and UDI-PI)
- Packaging UDI (Note: This is not an official term used in the EU MDR and IVDR, but we’re using it to help explain the concept. The Packing UDI is part of the UDI itself.)
1. Basic UDI-DI
The Basic UDI-DI identifies the device group that a particular device fits into. A device group is a group of products that all share the same intended purpose, risk class, essential design, and manufacturing characteristics. A device group is generally classified by medical device manufacturers as a “Product Family” or “Product Category,” depending on the internal nomenclature used within the company. The Basic UDI-DI functions as a parent or higher-level descriptor of a device.
NOTE: There can only be one Basic UDI-DI per UDI-DI.
The Basic UDI-DI is not printed on the product itself or on the packaging of a product, but rather it must be included in the following documents and applications:
- Certificates (Including Certificate of Free Sale)
- EU Declarations of Conformity
- Techical Documentation
- Summary of Safety and Clinical Performance
2. UDI (UDI-DI and UDI-PI)
The second component is the UDI itself, which consists of two parts:
Device Identifier (DI)
Production Identifier (PI)
The UDI-DI (Device Identifier DI, also referred to as “static”) identifies specific, detailed information about a particular device. If any of the below details should change, the device will need a new UDI-DI.
- Name or trade name of the device
- Device version or model
- If labelled as a single use device
- Packaged as sterile
- Maximum number of uses
- Need for sterilization before use
- Quantity of devices provided in a package
- Critical warnings or contra-indication
- CMR/endocrine disruptors
NOTE: There can be several UDI-DIs for one Basic UDI-DI.
Meanwhile, the UDI-PI (Production Identifier PI, also referred to as "dynamic") contains manufacturing information (including serial number, lot/batch number, software identification, and manufacturing or expiry date or both types of dates.)
To better illustrate this concept of Basic UDI-DI and UDI (UDI-DI and UDI-PI), let’s use a syringe as an example. The Basic UDI-DI would identify the category of a syringe, for example, "Enteral (Oral) Syringe."
A 5ml Enteral (Oral) Syringe – Sterile (Color: Purple) would get a unique UDI-DI and a 10m Enteral (Oral) Syringe – Sterile (Color: Orange) would get a unique UDI-DI. Both products would be associated to the same Basic UDI-DI. In this case, the "Enteral (Oral) Syringe," which defines the category.
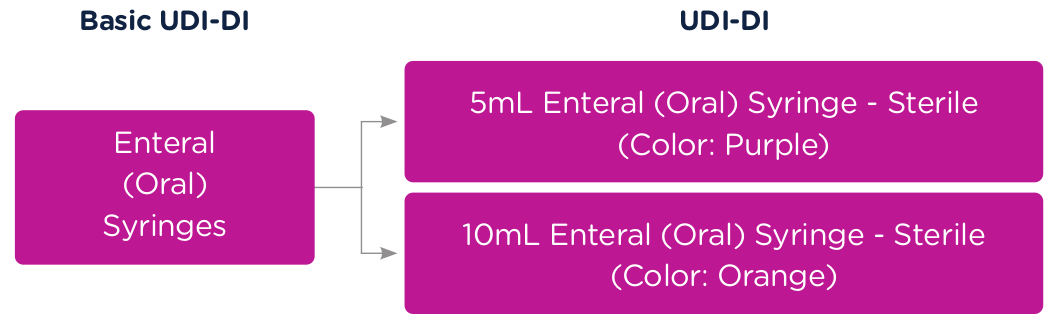
Each time that 5ml Enteral (Oral) Syringe – Sterile (Color: Purple) is manufactured at the same revision, it will get a new UDI-PI per lot. See the graphic below.

Each product is identical and therefore has the same UDI-DI. However, the UDI-PI changes to reflect the manufacturing date, lot number, expiry date, and serial number, as applicable.
The UDI will contain all device-specific information and have the same functions as the comparable database (GUDID) of the United States FDA. The main difference (in EUDAMED) is that the UDI data is divided into components of Basic UDI-DI, UDI, and Packaging UDI.
3. Packaging UDI
The third component of UDI is the Packaging UDI. (Note: This is not an official term used in the EU MDR and IVDR, but we’re using it to help explain the concept.)
Each level of packaging, except shipping containers, must receive its own unique UDI. Packaging UDI refers to the unique UDI assigned to higher levels of packaging instead of the device itself.
In the event of significant space constraints on the unit of use packaging, the UDI Carrier may be placed on the next higher packaging level.
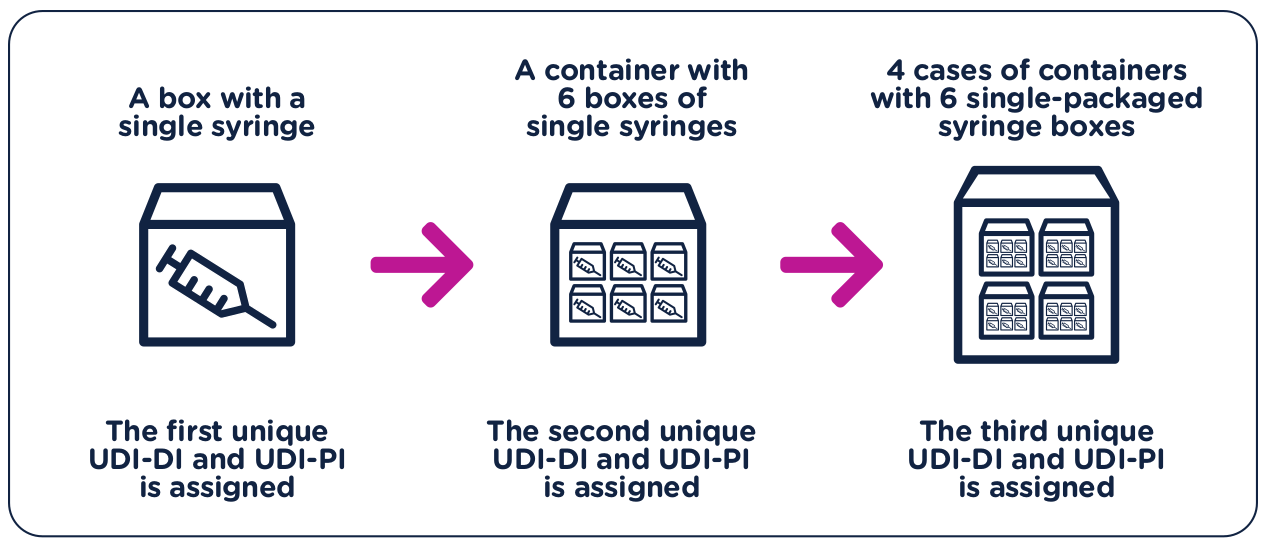
Returning to our earlier example of syringes, if a manufacturer first packages a single sellable syringe into an individual box, this package would receive its own UDI-DI and UDI-PI.
If then the manufacturer packages those individual boxes into containers of six (6), those containers would receive their own UDI-DI and UDI-PI.
And finally, if the manufacturer packages those six (6) containers into cases of four (4), those cases would receive their own UDI-DI and UDI-PI.
Each of those levels of packaging must be assigned its own UDI-DI and UDI-PI. The initial syringe did not change, but the way it is packaged did, therefore, requiring its own UDI-DI and UDI-PI.
Element 2: Placing UDI on the device and/or packaging
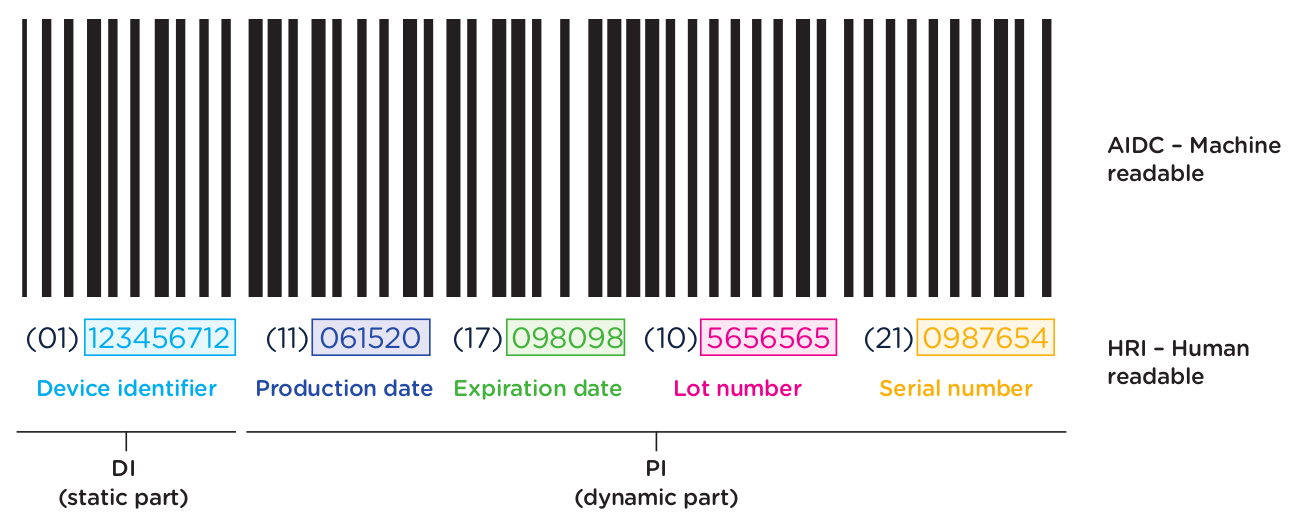
The second element to the UDI system is the placing of the UDI on the device or on its packaging through what is referred to as a “UDI Carrier.” The UDI Carrier is the part of the label that contains the UDI information that is applied directly to the device or included on the device packaging. The UDI Carrier should have both a machine-readable portion (AIDC) and a human-readable portion (HRI). (Specific details about each element of the UDI will be covered in Chapter 2.)
- Machine-readable form – AIDC – (Automatic Identification and Data Capture) is a barcode or other machine-readable technology that can be accessed automatically by scanning the UDI information.
- Human-readable form – HRI – (Human Readable Interpretation) is the numeric or alphanumeric code, which can be manually entered into the system for access to the UDI information.
If there are space constraints limiting the use of both the AIDC and HRI on the label, then only the AIDC is required to appear. However, on devices that are intended to be used in home-health care or other non-medical facility settings, the HRI would be required to appear.
Single-use devices may contain the UDI Carrier on its lowest level of packaging rather than on the device itself.
Reusable devices must include the UDI Carrier on the device itself, unless any type of direct marking would interfere with the safety or performance of the device, or if it is not technologically feasible to directly mark the device. If so, this should be properly documented in your design history file.
Most importantly, the UDI Carrier must be readable for the intended lifecycle of the device.
Below is an example of a GS1 AIDC and HRI barcode label.
Element 3: Storage of UDI information by Economic Operators
Storage of UDI information by "Economic Operators" is the third element of the UDI system. 2017/745 Articles 2(35), 22(1), and 22(3) define an economic operator as:
- A manufacturer
- An authorized representative
- A distributor
- An importer
- An investigator for clinical investigations
- A person who sterilizes systems or procedure packs
Class III, implantable device:
According to EU MDR 2017/745 Annex II, the manufacturer shall keep an updated list of all UDIs that it has assigned. Economic operators and all health institutions are required to store, preferably by electronic means, the UDI of all the devices for which they have supplied or with which they have been supplied.
For Devices Other than Class III:
Member States are encouraged, and in some cases require, health institutions to store, preferably by electronic means, the UDI of the devices with which they have been supplied. The UDI must also be included in any field safety notice for reporting serious incidents and field safety corrective actions.
The EU MDR and EU IVDR also give the European Commission authority to make additional requirements regarding the submission or maintenance of UDI information. In making those decisions, the European Commission must consider six (6) areas:
- Confidentiality and data protection
- Risk-based approach
- Cost-effectiveness of the additional measures
- The need to avoid duplications in the UDI system
- The needs of the healthcare systems of the member states
- Harmonization with other medical device identification systems
To continue reading this eBook including information about the EUDAMED database, UDI format requirements and issuing entities, implementation timelines, and key differences between the EU and US UDI systems, please register to download the full version



AI Agents and the Confidence Shift Inside MedTech IT
In some MedTech IT planning meetings, a new kind of confidence has started to show up.
Not everywhere. Not in every organization. But often enough that it is worth paying attention to.
It is subtle. Casual. The kind that appears when something new begins to feel inevitable
A VP of IT or a CIO sits in a planning meeting. Someone pulls up a demo. An AI agent drafts a regulatory summary, generates a workflow, and scaffolds an integration. It looks impressive. It is impressive
Then someone says it:
Why are we paying for a platform when we could build this ourselves?
I understand the impulse.
SaaS valuations are volatile. Boards are pressing on efficiency. Hiring is under scrutiny everywhere. AI arrives, and suddenly there is a clean story. Automate friction. Avoid headcount growth. Modernize everything
Some of that is real.
I am optimistic about AI. In the right hands, it is a genuine superpower
But hope, cost pressure, aggressive marketing, and very human psychology are colliding right now. That collision is shaping how executives talk about technology strategy
In regulated industries, that matters.
The Confirmation Bias Problem
When leaders already feel pressure to reduce costs or flatten organizations, they naturally gravitate toward stories that validate those instincts. Flashy demos and headlines about agents replacing departments reinforce the belief that a breakthrough must be right around the corner
Once that belief sets in, messy operational details get discounted. Risk gets deferred.
That does not make the technology fake.
It does explain why ambition so often outruns delivery reality
For CTOs and Regulatory leaders, this is the moment to slow the conversation down.
Because prototypes are not platforms.
What AI Actually Changes
Years ago, Harvard Business Review wrote about the “hidden data factory,” the idea that organizations accumulate thousands of small one-off efforts to clean data, reconcile systems, patch workflows, and keep operations moving. No single fix ever justifies a major initiative. In aggregate, it quietly costs millions
That concept maps directly to what AI is good at today.
Inside engineering organizations, we call this work toil.
The repetitive, manual, low-judgment effort that keeps systems running but should not consume the time of highly trained people. Environment setup. Data reconciliation. Migration scripts. Test generation. Documentation drafts. Classification lookups. Compliance artifacts
AI is excellent at eliminating toil. It removes friction, collapses queues, and gives teams back time
In regulated environments, that is meaningful.
But here is the distinction that matters:
Eliminating toil does not eliminate accountability
It does not remove the need for architecture, UX design, validation strategy, regulatory interpretation, or operational ownership.
What it does is allow smaller, more senior teams to focus on the work that actually differentiates platforms.
That is very different than from saying agents replace the platforms themselves

Why MedTech Regulatory Teams Are Delegating EUDAMED to IT
And Why That Creates Bigger Problems Over Time
As EUDAMED implementation accelerates and the UDI/Devices module becomes mandatory in May of 2026, many MedTech companies have made a seemingly practical decision. They hand EUDAMED compliance to IT.
At first glance, the logic feels sound. EUDAMED is a system. It requires integrations, data transmission, and technical connectivity. IT already owns those capabilities, so the project lands there.
But this handoff reveals a deeper misunderstanding of what EUDAMED actually represents. It is a tool that enables manufacturers to meet ongoing regulatory obligations that touch product data, submissions, post-market activities, and lifecycle management. EUDAMED also enables manufacturers’ ACTOR partners like Notified Bodies, Authorized Representatives, Importers, and Distributors to meet their obligations under those EU regulations. Treating it as an isolated, one-time IT project creates risks to EU regulatory compliance that grow and spread across partners over time. MDR/IVDR regulatory compliance cannot be established and maintained with a one-time technical integration.
The first problem with delegating EUDAMED to IT is what it signals internally. It frames the regulation as a single event rather than a continuous program.
EUDAMED is not just about getting data into a database. It requires ongoing updates tied to regulatory changes, product modifications, vigilance activities, certificates, and market status. Every change across the product lifecycle can trigger downstream updates in EUDAMED.
When EUDAMED is positioned as a one-time event, organizations underestimate the scope, effort, and ownership required to maintain compliance over time. That gap does not show up immediately. It appears months later when updates are missed; data falls out of sync, or responsibilities become unclear.
IT teams often take on EUDAMED with the expectation that once the pipes are built, the work is largely done. In reality, the opposite happens.
As regulatory data changes, IT becomes the default escalation point for updates they do not own and cannot validate. They are asked to manage regulatory timelines, interpret data requirements, and support continuous updates that fall outside their core mandate.
This creates friction on both sides. Regulatory teams feel blocked by technical dependencies. IT teams feel burdened by compliance work they were never meant to manage. Over time, updates slow down, workarounds emerge, and risk quietly increases.
The most damaging consequence of delegating EUDAMED to IT is architectural. When EUDAMED operates outside of a centralized Regulatory Information Management system, organizations lose the opportunity to reuse data and reduce burden across the business.
Most of the data required for EUDAMED already exists within product information management and resource planning systems. Product registrations, certificates, submissions, UDI, and post-market data are not new. They are part of the regulatory lifecycle. When EUDAMED is disconnected from RIM, teams are forced to duplicate work, reconcile inconsistencies, and manually manage updates across systems.
Instead of becoming a natural extension of regulatory operations, EUDAMED turns into another silo. One that increases workload rather than streamlining it.
Establishing and maintaining regulatory information in EUDAMED is a regulatory obligation, not a technical one. While IT plays a critical role in enablement and integration, there should be a strong partnership between regulatory and IT (or a third-party submitter), but IT shouldn’t own it completely.
When EUDAMED is managed as part of a centralized RIM approach, organizations gain consistency, traceability, and reuse. Regulatory teams can leverage existing data, control updates at the source, and reduce the ripple effects of change across departments. IT supports the infrastructure, but regulatory owns the process.
This shift also changes how organizations think about compliance. Instead of reacting to EUDAMED as a standalone requirement, they treat it as part of a broader regulatory operating model that supports long-term compliance and growth.
Delegating EUDAMED to IT is rarely a conscious strategy. It is usually a symptom of fragmented regulatory operations and unclear ownership.
As MedTech companies scale globally and regulatory expectations continue to evolve, these handoffs become harder to sustain. EUDAMED exposes the cost of treating regulatory compliance as a series of isolated projects rather than an ongoing operational discipline.
The companies that navigate EUDAMED successfully are not the ones with the most complex integrations. They are the ones that anchor EUDAMED within regulatory operations, supported by centralized RIM systems that establish data consistency and reduce duplication, improve visibility, and spread the burden across the organization in a controlled way.

Agentic AI and the Future of Regulatory Operations
Why Regulatory Operations Is Ready for Agentic AI
Regulatory operations teams are under increasing pressure. Global regulatory complexity is rising, data volumes continue to grow, and teams are expected to move faster, often without additional headcount. At the same time, employee turnover and fragmented systems make it harder to maintain continuity and institutional knowledge.
As outlined in the RIM & AI Maturity in MedTech Executive Guide, many organizations are still operating with scattered regulatory data, reactive processes, and manual workflows. These conditions increase compliance risk and slow growth.
This environment has created the conditions where a more advanced form of AI can deliver meaningful value. That is where agentic AI comes into play, not as a replacement for regulatory expertise, but as a way to strengthen how regulatory operations function day to day.
What Is Agentic AI and Why It Matters
Most AI used in regulatory environments today is assistive. It helps classify documents, extract text, or answer questions when prompted. Agentic AI goes further by operating within defined workflows and processes.
Agentic AI systems can monitor structured regulatory data continuously, identify upcoming risks or deadlines, recommend actions based on rules and historical context, and surface next steps within governed processes. Instead of responding to requests, agentic AI supports execution by working alongside regulatory teams inside their operational systems.
The distinction is important. In regulated environments, value does not come from generative output alone. It comes from intelligence that is embedded, auditable, and aligned with how regulatory work actually gets done.
Moving Regulatory Teams Off the Data Treadmill
The executive guide describes early-stage regulatory teams as being stuck on a back-office data treadmill. Highly skilled professionals spend a disproportionate amount of time searching for information, reconciling spreadsheets, and repeating manual tasks rather than applying their expertise strategically.
Agentic AI helps reduce this burden by continuously organizing and validating regulatory data, identifying missing metadata or inconsistencies early, and reducing reliance on individual memory or tribal knowledge. Over time, this improves not just efficiency, but operational resilience. Teams become less vulnerable to audits, turnover, and last-minute regulatory surprises.
Why Agentic AI Depends on Operational Maturity
One of the most important insights from the paper is that AI value scales with RIM maturity. Advanced AI capabilities are not effective without centralized regulatory information and standardized processes .
At higher maturity levels, AI can surface upcoming risks across markets and renewals, analyze submission history to recommend reusable content, and identify bottlenecks before they impact timelines. At this stage, agentic AI begins to function as an operational partner, helping teams anticipate issues rather than react to them.
This is also where many organizations encounter friction. Skipping foundational steps may create the appearance of progress, but it limits reliability and long-term impact. Agentic AI is only as effective as the data, governance, and workflows it operates within.
From Task Automation to Predictive Compliance
At the most mature stage of regulatory operations, AI becomes fully embedded in daily work. The guide describes this level as one where real-time monitoring, predictive analytics, and continuous improvement are standard practice .
In this environment, agentic AI supports predictive compliance by identifying emerging risks, highlighting resource constraints, and improving visibility across submissions and renewals. These insights allow teams to act earlier and with greater confidence.
The paper is clear on one point. AI enhances regulatory expertise, but it does not replace it. Human judgment remains essential for interpretation, decision-making, and accountability. The real value of agentic AI is that it frees regulatory professionals from low-value work so they can focus on the decisions that matter most .
Regulatory Operations as the Heart of Compliant Growth
The most significant impact of agentic AI is not automation alone. It is the elevation of regulatory operations from a reactive support function to the heart of compliant growth.
Organizations that invest in strong RIM foundations, data governance, and workflow integration are better positioned to apply AI in a way that is safe, scalable, and durable. When implemented thoughtfully, agentic AI helps regulatory operations keep pace with growth, reduce risk, and support faster, more confident decision-making across the business.
The Future of MedTech Compliance: How AI Is Transforming Regulatory Affairs
MedTech regulatory affairs teams are facing a turning point. Regulations are expanding in number and complexity, resources are limited, and manual processes cannot keep up. At the same time, artificial intelligence (AI) has become a serious topic of discussion in regulatory circles. Leaders are beginning to ask: How can AI help us manage change, reduce risk, and accelerate compliance efforts?
The answer is clear: AI is no longer just a buzzword. When combined with effective regulatory information management (RIM), it can be a powerful enabler of efficiency, accuracy, and strategic decision-making.
Why AI is Trending in Regulatory Affairs
The Surge of Regulatory Data
Regulatory teams must now track requirements from multiple global markets. Each regulator frequently updates its regulations, guidances, templates, and recognized standards, which creates large volumes of data to organize and analyze. AI can scan and classify this information, highlight changes, and prepare it for structured use within RIM systems.
Doing More with Limited Resources
Most teams are expected to deliver more without additional staff. High turnover makes continuity difficult, and according to the 2024 RAPS Global Workforce Report, the number of professionals “open to work” has grown in North America and Europe. AI offers relief by taking on repetitive tasks such as document formatting or data entry, allowing experts to focus on higher-value work.
Global Complexity and Diverging Standards
No two markets are exactly alike. AI can help by flagging differences, surfacing potential risks, and recommending reusable content drawn from a company’s submission history. Faster, more accurate submissions directly improve time-to-market and compliance outcomes.
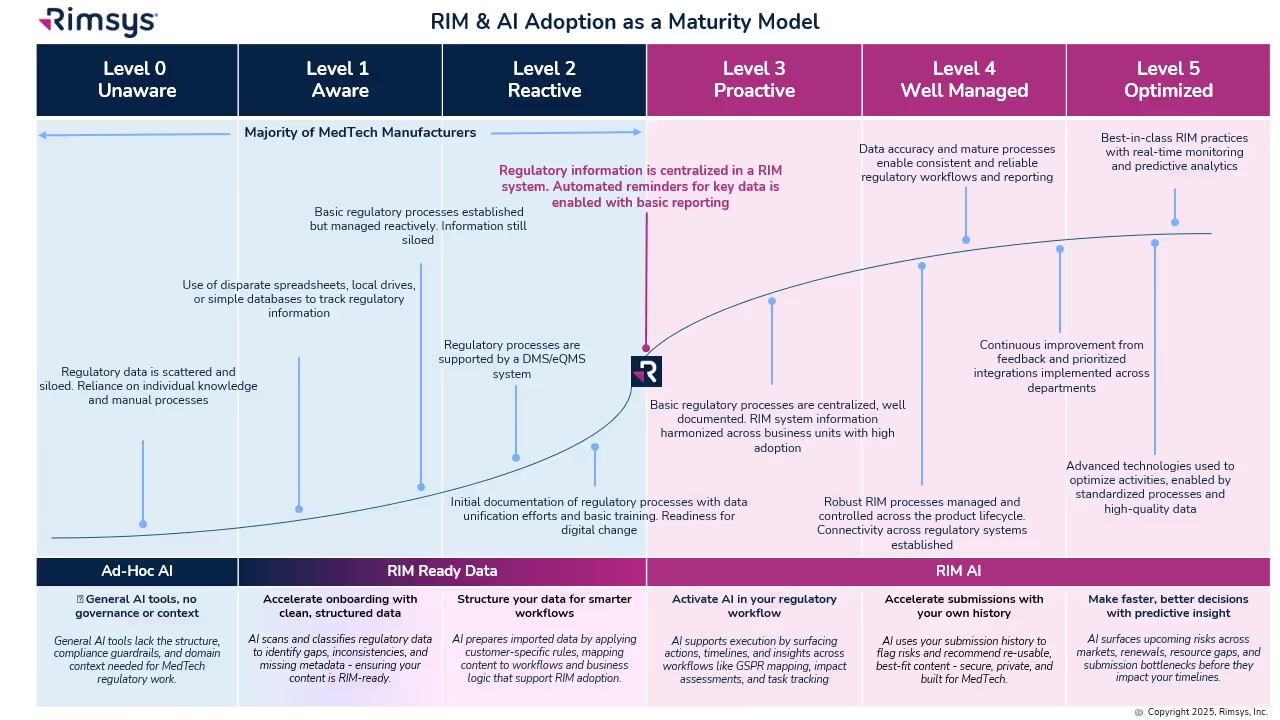
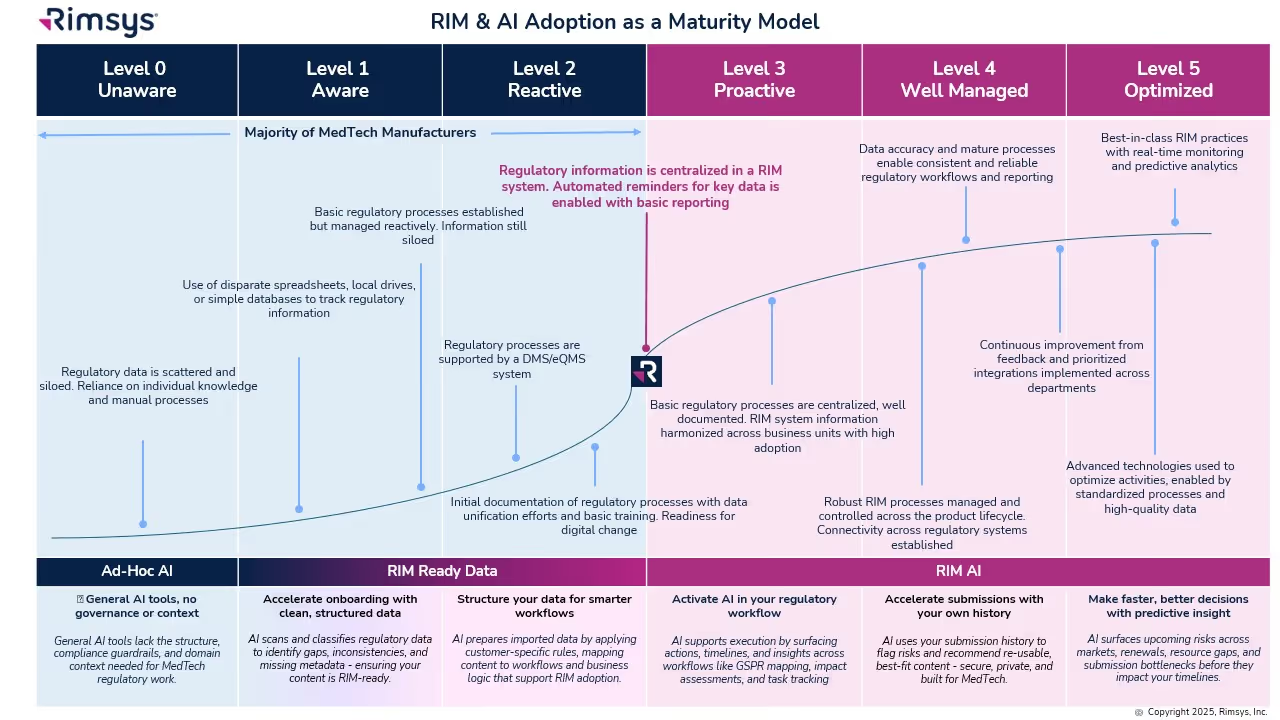
The RIM & AI Adoption Maturity Model
Not every organization is ready to fully embrace AI. Success depends on RIM maturity: how structured and centralized your regulatory processes and data are. The RIM & AI Adoption Maturity Model provides a roadmap from basic to optimized states.
- Levels 0–2: Early Stage
- Data is siloed and processes are ad hoc. AI provides value in isolated ways, such as cleansing records or scanning for regulatory changes.
- Level 3: Proactive
- A RIM system centralizes information. AI begins to surface reminders, deadlines, and global impact assessments.
- Level 4: Well Managed
- Processes are standardized across the lifecycle. AI generates insights, monitors KPIs, and supports reuse of regulatory content.
- Level 5: Optimized
- AI is fully embedded, delivering predictive analytics, continuous monitoring, and smarter decision-making.
Practical Applications of AI Today
Today, regulatory teams see the greatest opportunities in:
- Regulatory submissions: Automatically detecting changes in templates and suggesting updates.
- Document classification: Using natural language processing to tag and organize regulatory documents.
- Regulatory intelligence: Monitoring health authority updates and highlighting what matters most.
- Impact assessments: Linking changes (e.g., regulations/standards/design) directly to the affected products and registrations and evaluate the potential impact.
- Content reuse: Recommending approved content to accelerate submissions.
How to Start Your AI Journey in Regulatory Affairs
Adopting AI is not about jumping to the most advanced capabilities overnight. Instead, consider these steps:
- Assess your RIM maturity. Where does your organization sit on the 0–5 scale? What foundational gaps (data centralization, process standardization) need to be addressed first?
- Identify quick wins. Focus on repetitive, rules-based tasks where AI can add value without major disruption.
- Implement governance. Establish policies for safe, compliant AI use, particularly around data privacy and model training.
- Pilot in phases. Start small, validate results, and expand AI use as confidence and maturity grow.
- Keep people at the center. AI should enhance the expertise of regulatory professionals, not replace it.
Building a Smarter Future for MedTech Compliance
AI is becoming a trending topic in regulatory affairs not just because it’s new, but because it directly addresses the challenges teams face: rising complexity, limited resources, and scattered data.
For organizations that take this approach, the benefits are clear: lower compliance risk, faster execution, and stronger competitive positioning. AI does not replace regulatory professionals. Instead, it enables them to spend less time on manual tasks and more time on strategic contributions that improve patient access to life-changing technologies.
In other words, AI isn’t about futuristic transformation. It’s about helping regulatory teams step off the “data treadmill” and reclaim their time for what matters most: bringing safe, life-changing medical technologies to patients faster.
.avif)
Rimsys Becomes the Trusted Regulatory Partner for 6 of the Top 12 Global MedTech Manufacturers
“Adoption by half of the top global MedTech manufacturers is a powerful validation that we’re not just a solution, we’re setting the new gold standard for regulatory excellence.”
Pittsburgh, PA - August 7, 2025 - Rimsys, the leading Regulatory Information Management (RIM) software purpose-built for the MedTech industry, today announced a significant milestone: 6 of the world’s top 12 medical device manufacturers now rely on Rimsys to manage and streamline their global regulatory operations.
This milestone further solidifies Rimsys’ position as the trusted partner to the world’s most innovative and quality-focused MedTech companies.
Click here for the full list of the top 12 global MedTech companies.
“Today’s regulatory environment demands more than spreadsheets. Leading manufacturers recognize that regulatory operations are mission-critical, revenue-generating departments and need systems to match that level of importance,” said James Gianoutsos, Founder and CEO of Rimsys.
Rimsys’ unified, enterprise-grade RIM platform centralizes and automates critical regulatory processes—including market registrations, Unique Device Identification (UDI), essential principles/GSPR, and submissions management—reducing compliance risk and accelerating market access. Specifically tailored to the needs of medical device and diagnostics companies, Rimsys enables seamless collaboration across RA, QA, and commercial teams while delivering the audit-ready transparency global regulators demand.
“As more organizations embrace regulatory digital transformation, Rimsys is proud to lead the industry forward,” added Gianoutsos. “Adoption by half of the top global MedTech manufacturers is a powerful validation that we’re not just a solution, we’re setting the new gold standard for regulatory excellence.”
To learn more about the Rimsys, please visit www.rimsys.io.
About Rimsys
Rimsys is the leading provider of Regulatory Information Management (RIM) software purpose-built for MedTech manufacturers. The comprehensive platform digitizes and automates regulatory activities, helping MedTech regulatory affairs teams to efficiently achieve regulatory compliance and get products to market faster. Rimsys is designed around MedTech workflows and supports a full breadth of regulatory functions including registrations, submissions, UDI, EUDAMED compliance, essential principles, and standards management in a unified platform. Rimsys is trusted by half of the world’s top 12 MedTech companies to power their global regulatory operations. For more information, visit www.rimsys.io.
%2520(855%2520x%2520268%2520px).avif)
Rimsys Announces Bulk UDI Submission and Rimsys Connect™ to Empower MedTech Regulatory Teams
New solutions deliver enterprise-grade data access and streamlined EUDAMED compliance, driving smarter, faster decisions across the business
Pittsburgh - April 29th, 2025 - Rimsys, the global leader in Regulatory Information Management (RIM) software for the MedTech industry, today announced two major enhancements to its platform: expanded Unique Device Identification (UDI) capabilities to support EUDAMED machine-to-machine (M2M) bulk transmission and Rimsys Connect™, a new enterprise Change Data Capture (CDC) solution that provides near real-time synchronization of Rimsys data with customers’ Business Intelligence (BI) solutions.
Together, these capabilities are designed to help MedTech organizations streamline compliance, reduce manual effort, and unlock the full strategic value of their regulatory data.
New UDI Capabilities Support EUDAMED Readiness
The UDI enhancements extend Rimsys’ industry-leading Universal UDI® framework, enabling MedTech teams to manage complex, global UDI programs in one unified RIM system. Key new capabilities include:
- Approving multiple records simultaneously via a simple, scalable workflow
- EU data governance support with all required attributes for EUDAMED transmission
- Bulk submission of records to both the GUDID and EUDAMED databases
These features allow teams to eliminate time-consuming, record-by-record processing, helping them meet the mandatory January 2026 EUDAMED compliance deadline with confidence.
“We’ve partnered closely with our customers to develop a UDI offering that meets increasing regulatory complexity and is easily scalable as new regulations come online,” said Adam Price, Director of Regulatory and Technical Programs at Rimsys. "We’re not only giving customers the ability to meet EUDAMED compliance but enabling them to manage their global UDI program in a single-sourced RIM solution for complete visibility.”
Introducing Rimsys Connect™: Enterprise Data Access, Redefined
Rimsys Connect™ offers enterprise customers a powerful new way to leverage regulatory data across the business. Built on a scalable, event-driven architecture, it provides secure, structured, near real-time streaming of Rimsys data into any modern data warehouse solution—such as Snowflake, Amazon S3, and Salesforce Bulk API 2.0.
“Rimsys Connect™ is not just a connector—it’s a strategic enabler,” said James Gianoutsos, Founder and CEO of Rimsys. “We’re giving regulatory affairs teams the ability to deliver insights that influence launches, accelerate tender responses, and align compliance with business impact. With Connect, RA teams become true strategic partners.”
By providing full access to customer data—registrations, UDI, projects, tasks, and custom attributes — Rimsys Connect™ supports a wide variety of enterprise use cases with customers’ own business intelligence solutions:
- Tracking on-time submission and decision KPIs
- Aligning registration timelines with product launch dates
- Conducting ROI analysis for renewals and market prioritization
- Accelerating tender readiness by combining RIM and PLM data
- Supporting post-market surveillance dashboards
While the initial release will focus on data access, Rimsys plans to expand Connect with curated BI templates and best practices to further accelerate enterprise customer time-to-value.
Solving the Data Fragmentation Problem for MedTech
Many regulatory affairs teams remain constrained by outdated tools, fragmented data sources, and increasing demands to deliver strategic insights to executive and commercial stakeholders. Rimsys Connect™ addresses these challenges by eliminating manual reporting workflows and enabling teams to analyze their regulatory data alongside financial, marketing, and quality systems.
“With Rimsys Connect™, regulatory teams can visualize and analyze their data in real time, assess launch readiness, and deliver more value to their organizations. This is how RA becomes a catalyst for better decisions—not just compliance,” said Gianoutsos.
Both Rimsys’ expanded UDI capabilities and Rimsys Connect™ will be available this summer. Those interested in learning more about these solutions and how they will enable greater automation, efficiency, and compliance can visit our booth at RAPS Euro Convergence May 13-15 in Brussels, Belgium, or sign up for Rimsys’ product update webinar on Thursday, May 22nd at 10 AM ET.
Read the press release here.
About Rimsys
Rimsys is improving global health by accelerating delivery and increasing availability of life-changing medical technologies. Rimsys Regulatory Information Management (RIM) software digitizes and automates regulatory activities, helping MedTech regulatory affairs teams to plan more effectively, execute more quickly, and confidently ensure global regulatory compliance. Rimsys is designed around MedTech workflows and supports a full breadth of regulatory activities including registrations, submissions, UDI, essential principles, and standards management in a unified platform. For more information, visit www.rimsys.io.
Contacts:
marketing@rimsys.io
