


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.
.avif)

2023 Regulatory performance report
Today at Rimsys, we unveiled the 2023 MedTech Regulatory Performance Report, a new set of insights into the state of medtech regulatory affairs. Compiled based on interviews with 200 regulatory professionals and executives, the study provides a detailed look into how regulatory teams are staffed, their processes, the tools they use, and ultimately how they perform.
Why did we create this study? There were two driving factors behind the research. The first was a common theme that we heard from a number of our customers: Regulatory leaders don’t have clear data and benchmarks. They don’t necessarily know how long a new market submission should take, and how to plan for or assess the work of their teams. While other studies look at the medtech industry broadly or the state of the regulatory profession, this study tries to build a comprehensive resource for regulatory (and company) leaders.
The second factor was really for ourselves and the team at Rimsys. As a company building solutions specifically for medtech regulatory affairs, we wanted more insight into where companies were successful, where they struggled, and where we can add value.
What did we find? Regulatory teams perform a lot of hero work and rate themselves highly for their accomplishments. At the same time there is a lot of opportunity for process improvements, and companies that invest in digital transformation for regulatory affairs see better performance.
Regulatory professionals are superheroes
Regulatory teams are generally pretty small. Most companies have less than 10 full-time regulatory professionals. These small teams complete an enormous amount of work. Last year on average, RA teams completed 50 license renewals, 50 license updates, and 10 new market submissions. This is impressive output.
Digging a bit under the covers, we found that this output relied heavily on the support of external consultants. 90% of companies use consultants to keep pace with their regulatory workload. Front-line employees also struggle with burnout. They were much more likely to report feeling under-resourced than regulatory leaders.
But process problems persist
A lot of regulatory work remains extremely manual. 70% of regulatory teams spend half their time or more on repetitive administrative tasks. All of this manual work increases the frequency of errors and required rework. 61% of companies reported a major non-compliance incident in the past 2 years.
Manual work also makes it difficult to complete regulatory projects in a timely fashion. Teams completed a lot of projects, but each took a long time. Over half of all companies spend 4 months or more on license renewals, license updates, and new market submissions.
Moving regulatory affairs forward
As regulatory requirements become more complex, there’s a natural question about how teams will work moving forward. MDR & IVDR in Europe have significantly increased the regulatory workload required to bring and keep products on the market. Will organizations be able to keep pace with the same resources, tools, and processes?
No, and the performance report shows that medtech companies are investing to improve their regulatory capabilities. The majority of companies are planning to increase the sizes of their RA teams in 2023, and 40% expect to increase their investments in regulatory software. Companies are increasingly adopting specialized software to better support regulatory processes.
Dig into the survey results
The full survey results provide insights into more aspects of regulatory performance. They show that companies need to take a deeper look into their processes and how regulatory resources are allocated. There are two ways to learn more:
- Visit the survey page to see the full results (the survey whitepaper can be downloaded at no cost)
- Watch the recording of our webinar with PA Consulting. We discuss the survey results in more detail and share our regulatory predictions for 2023



Post-market surveillance for medical devices in the European Union
This article is an excerpt from Post-market surveillance for medical device in the European Union.
Table of Contents
- What is post-market surveillance?
- What classes of medical devices require post-market surveillance?
- Components of a successful post-market surveillance plan
- PMS data requirements
- Post-market surveillance system goals
- Required post-market surveillance reporting
- Embracing post-market surveillance as an integral part of your quality program
- Getting started with post-market surveillance
Post-market surveillance (PMS) is designed to monitor the performance of a marketed medical device by collecting and analyzing field use data. Article 10 of the EU MDR and IVDR requires all device manufacturers to have a post-market surveillance system in place. The main elements of the PMS are laid out in Article 83, and additional details for lower-risk and higher-risk devices are covered in articles 84 and85, respectively.
In general, a PMS system consists of both proactive activities and reactive, or vigilance, activities. While post-market surveillance and vigilance are sometimes used interchangeably, vigilance consists of separate activities that feed post-market surveillance programs.
Post-market surveillance systems are used to collect and analyze data not only about the manufacturer’s device but also about related competitors’ devices that are on the market. Data collected through PMS procedures is then used to identify trends that may lead to, among other things, quality improvements, updates to user training and instructions for use, and identification of manufacturing issues.
Note that “market surveillance” encompasses activities performed by a Competent Authority to verify MDR compliance, and should not be confused with the topic of this ebook,“post-market surveillance," which is performed by the manufacturer.
All medical devices marketed in the EU require some level of post-market surveillance, and all medical device manufacturers must implement a post-market surveillance system (PMS). The requirements of the PMS, however, vary and should be “proportionate to the risk class and appropriate for the type of device” (MDR Chapter VII). In particular, the type and frequency of reporting vary based on a device’s risk class.
A post-market surveillance plan (PMS) is an integral part of a manufacturer’s quality management system and provides a system for compiling and analyzing data that is relevant to product quality, performance, and safety throughout the entire lifetime of a device. The PMS should also provide methods for determining the need for and implementing any preventative and corrective actions. A PMS system should include and define:
Surveillance data sources
With the increased focus on proactive risk identification in the MDR, it is important to design post-market surveillance systems that actively acquire knowledge and detect potential risks. It is not sufficient to rely solely on spontaneous reporting by healthcare providers, patients, and other stakeholders.
In addition to information coming from Clinical Evaluation Reports and complaint and adverse event reporting, typical sources of surveillance data include:
• Social media networks: Because many of your stakeholders may be communicating on social media networks, it is important to employ social listening techniques and/or tools to identify issues and concerning trends as they develop.
• Industry and academic literature: Any studies, academic papers, and other literature that addresses similar devices or the specific use cases for which your device is designed should be evaluated. In particular, risk factors and adverse events identified with similar devices should be closely examined. It is also important to identify newer technologies that may affect the benefit-risk ratio and establish a new definition of “state of the art” for the device type.
• EUDAMED: While the European Database on Medical Devices (EUDAMED) is not yet fully functional, it is intended to provide a living picture of the lifecycle of all medical devices marketed in the EU. Manufacturers should take special care to consider information for similar devices made available through the EUDAMED system in the future.
• Registries: Patient, disease, and device registries can provide information that informs the clinical evaluation process which provides input into the post-market surveillance system.
Data analysis methodology
A well-defined data analysis methodology will accurately identify trends and lead to defendable decisions in the application of post-market experience. Once the necessary information has been identified and collected, and potentially cleaned of incomplete or otherwise unusable data, the data needs to be analyzed.
The goal is to identify meaningful trends, correlations, variations, and patterns that can lead to improvements in the safety and efficacy of the device. There are many data analysis tools available that can assist with:
• Regression analysis that will identify correlations between data (e.g. the device location/geography correlates to battery life).
• Data visualization that can be useful in spotting trends in the data.
• Predictive analytics, which can be particularly useful with large data sets, to identify future trends based on historical data.
• Data mining, which is also normally used with large datasets, to organize data and identify data groups for further analysis.
Benefit-risk indicators and thresholds
The MDR requires that medical device manufacturers not only demonstrate the clinical benefit of their device but also quantify the benefit-risk ratio. The benefit of a device must be shown to clearly outweigh the risk for it to gain market approval. Article 2 (24) of the MDR defines the benefit-risk determination as “the analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose when used in accordance with the intended purpose given by the manufacturer.”
A PMS system should clearly define benefit-risk calculations and the data used to support them. Post-market surveillance activities are critical in order to re-evaluate and maintain the benefit-risk calculations and determinations of a device throughout its life. Information that is gained through a PMS system can lead to:
• Identification of new risk factors.
• Adjustments to risk frequency and/or severity values based on actual use data.
• Adjustments to established risk calculations based on new “state of the art” technologies becoming available.
• Adjustments to established benefit calculations based on actual use data.
While complaint handling and other feedback tracking are more often described as part of post-market vigilance systems, they play a role in the more proactive post-market surveillance processes as well. A PMS system should define ...
To continue reading this ebook, download the full version.

Canada medical device regulations
Health Canada medical device regulations
Canada has one of the most stringent and well-respected regulation processes, not only for medical devices but for the overall health and safety of its citizens. Canada uses a risk-based approach to the regulation of medical devices, where the review before approval depends on the potential risk that the use of the device presents or could potentially present. Devices are categorized into four classes based on the risk, with Class I devices presenting the lowest potential risk (e.g. a tongue depressor) and Class IV devices presenting the greatest potential risk (e.g. a pacemaker). Class II, III, and IV medical devices must have a Medical Device License to be sold in Canada, while companies selling Class I medical devices in Canada are required to have a Medical Device Establishment License (MDEL).
The regulation of medical devices in Canada is driven by The Food and Drugs Act (R.S.C., 1985, c. F-27). This act includes food, drugs, cosmetics, and devices. The regulation specific to devices is the Medical Devices Regulations (SOR/98-282). The information within the regulations includes, but is not limited to, classification, manufacturer’s obligations, associated fees, labeling requirements, establishment license, incident reporting, recalls and much more. The most recent update to the regulation was Interim Order No. 3, Respecting the Importation and Sale of Medical Devices for Use in Relation to COVID-19.
Most countries have an established system in place for notifying the public (including manufacturers) of initiatives and actions that they plan to take to maintain and improve the safety of medical devices - and Canada is no different. As a department, Health Canada is responsible for administering Acts and Regulations, and for implementing government-wide regulatory initiatives. All the government's Acts and Regulations can be found on the Justice Canada website. Let’s look at some of their processes and review some of their initiatives and action plans through 2024.
Forward Regulatory Plan
Canada’s Forward Regulatory Plan is designed to share anticipated regulatory changes or actions. The forward regulatory plan gives consumers, businesses, and other stakeholders an opportunity to review and comment on anticipated changes. It should be noted though, that this forward regulatory plan can be modified at any point.
The Forward Regulatory Plan: 2022 - 2024 provides information about specific regulatory activities and initiatives that Health Canada aims to finalize through 2024. This plan sets up a timeframe for activities and initiatives to be completed. Pre-publication and final publication announcements are published in the Canada Gazette, the official newspaper of the Government of Canada.
There are currently several initiatives in this Forward Regulatory Plan including those that fall under the Consumer Product Safety Act, Hazardous Products Act, and Food and Drugs Act, to name a few. The following are major medical device initiatives included in the plan, which fall under the Food and Drugs Act.
- Amendments to the Fees in Respect of Drugs and Medical Devices Order (Natural Health Products Fee Proposal) [New 2022-04-01]
- Advanced Therapeutic Products Pathway for Adaptive Machine Learning-enabled Medical Devices [Updated 2022-10-01]
- Regulations Amending the Food and Drug Regulation, Natural Health Products Regulations and the Medical Devices Regulations (Modernization of the Regulation of Clinical trials) [Updated 2022-10-01] [Associated with the targeted Regulatory Review Sectoral Roadmap] [Associated with the stock review plan]
- Regulations amending the Medical Devices Regulations (Agile Regulations for Licensing Medical Devices) [Updated 2022-10-01] [Associated with the targeted Regulatory Review Sectoral Roadmap] [Associated with the stock review plan]
- Regulations Amending the Medical Devices Regulations (Interim Order No. 3 Respecting the Importation and Sale of Medical Devices for Use in Relation to COVID-19) [Updated 2022-10-01]
- Modernizing the Medical Device Establishment Licensing (MDEL) Framework [Updated 2022-10-01]
Stock Review Plan
The Stock Review Plan is a public list and description of planned reviews of existing regulations that Health Canada is proposing within a two-year period. As of the date of this article, the on-going and proposed reviews for medical devices include:
On-going Reviews:
- Regulations Amending the Medical Devices Regulations (Agile Regulations for Licensing Medical Devices), formerly referred to as Regulations Amending the Medical Devices Regulations (Agile Regulations)
- Modernizing Medical Device Establishment Licensing Framework including the recall of medical devices, formerly referred to as Modernizing Drug and Medical Device Establishment Licensing Frameworks – amendments to the Food and Drug Regulations and Medical Devices Regulations
Proposed new reviews:
Additional regulation updates
Regulatory Initiative Plan
Health Canada’s regulatory initiative agenda aims to provide more regulatory flexibility to support innovative research and health product development and to contribute to the country's biomanufacturing and life sciences strategy.
The 5 key pillars in the regulatory innovation agenda include:
- Modernizing clinical trial regulations
- Enabling advanced therapeutic products
- Agile regulations for licensing drugs
- Agile regulations for licensing medical devices
- Information to Canadians mobile strategy
UDI
Unique Device Identification (UDI) is a medical device requirement in most countries, but every country is at a different stage of implementing UDI requirements. So where does Canada stand with UDI? As of June 2021, Canada was exploring the feasibility of a UDI system, including options for development of a UDI database by using internal, existing infrastructures. They propose to develop this system based on the work and experience of the International Medical Device Regulators Forum (IMDRF), as well as the United States FDA’s UDI system, to help structure the principles and design of a UDI system for Canada. No further announcements have been made since the writing of this article.
COVID-19
February 2023, Canada announced a few updates relating to regulations about COVID-19 including:
- Amendments to Medical Devices Regulations to continue importation and sale of COVID-19 medical devices
- Medical devices for use in relation to COVID-19
- List of Medical Devices for an Urgent Public Health Need in Relation to COVID-19
Keep up to date with news and announcements from Health Canada by referring to our Medical Device Regulatory Market Profiles section on our website.
MDR transition example under the extension
Medtech manufacturers cannot delay the preparation for transitioning devices to MDR. While the final deadlines were pushed, this was done primarily to address notified body capacity issues. The MDR transition period extension for legacy devices does NOT allow manufacturers to delay/deprioritize efforts until the end of the applicable extension period since many activities must be fulfilled now to utilize the extension. Read 6 reasons medtech companies shouldn’t delay MDR certification for additional information.
To illustrate the timeline, we are going to look at an example of a Class IIb non-implantable device.
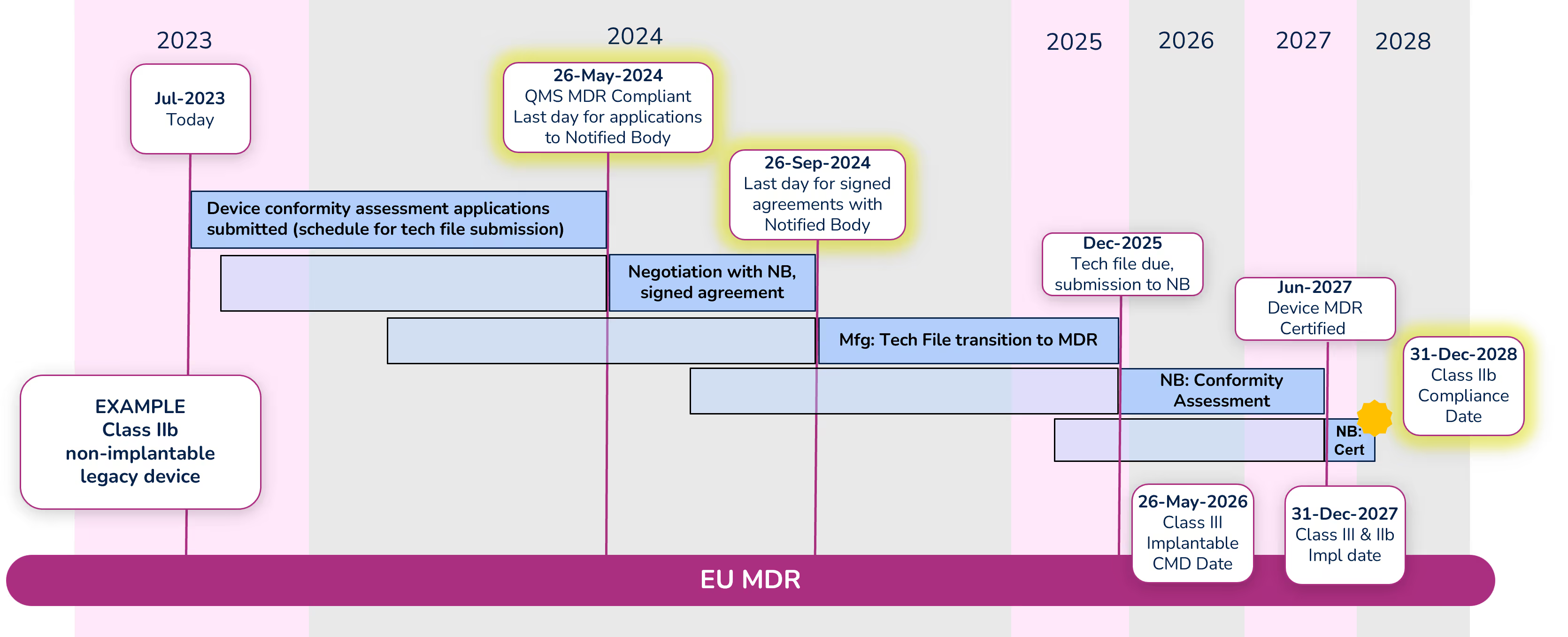
Note: Dates within the diagram for tech file submission and certification are based on your signed agreement with your Notified Body and the outcome of the conformity assessment. The dates shown are only an example.
Apply for conformity assessment - May 26, 2024
Even with the extension, a quality management system (QMS) that is compliant with MDR is required by May 26, 2024. Manufacturers must submit an application for the device conformity assessment to a Notified Body (NB) before that date as well. There are significantly fewer Notified Bodies that are certified to MDR, and due to their limited resources, manufacturers should begin working with their NB immediately if they have not already done so. Note that the device technical file does not need to be submitted with the application, but a submission schedule must be.
Sign agreement with Notified Body – September 26, 2024
Before signing an agreement with the Notified Body, expect them to counter your application with a new submission date. Manufacturers can negotiate the date with their Notified Body, but a signed agreement must be in place before September 26, 2024. While in this negotiation phase, keep in mind the MDR compliance deadline for the device (Class IIb in this example) is December 31, 2028, and work backward accounting for the various activities that must take place and their durations. It is extremely important to define a realistic schedule because not meeting that schedule will cause significant administrative complications and raise the potential risk that a device is not certified before the required deadline.
Conformity Assessment
The Notified Body will begin the conformity assessment once they receive the technical file for your device. Technical files should be submitted to the Notified Body by the date defined in your agreement (Dec 2025 in our example).
We are hearing that conformity assessments for many devices are taking 12-18 months. During that period, be prepared to answer questions and participate in conversations with your NB. In some cases, there can be significant back-and-forth between the manufacturer and the NB during the conformity assessment. Once the conformity assessment is complete (Jun 2027 in our example) it can take up to another 3-6 months for certificate issuance.
MDR extension reference documents
- Q&A Document for Regulation 2023/607 – Published by the EU Commission, this document answers some of the common questions around the extension in “plain English.”
- Regulation 2023/607 amending MDR (EU) 2017/745 and IVDR (EU) 2017/746 in regard to the transitional provisions and removal of sell-off periods for medical devices and for in vitro diagnostic medical devices.
- Notified Body Confirmation Letter template published by Team NB. This is a confirmation letter created by the manufacturer for the Notified Body to sign. The letter is not mandatory but is strongly recommended to provide objective evidence that the conditions of the extension have been met extending the validity of the CE certificate.
For more information, watch the replay of our recent "Ask Us Anything" webinar on the EU MDR Transition Period Extension.

Declarations of Conformity
What is a Declaration of Conformity?
A Declaration of Conformity (DoC) is a required self-certifying document created by the medical device manufacturer to state that a device or a series of devices complies with the conformity requirements of a particular country’s regulations. If a DoC is required in a country, specifically in the European Union, the DoC confirms that a product is in compliance with all relevant European product safety requirements. DoC is required for each product and identifies the party responsible for market authorization activities in the applicable country.
Why is a Declaration of Conformity necessary?
Declarations of Conformity require the signee to take responsibility for a product’s compliance within that country’s rules and regulations. It also allows government authorities and regulatory bodies to identify who is legally responsible for a product and its regulatory activity, such as obtaining a CE mark.
Why are Standards often listed in the Declaration of Conformity?
Governments often use industry standards to define sections of the conformity assessment for particular devices. These standards are developed by standards organizations, such as ISO and IEC. For example, the ISO 13485:2016 standard defines quality management system (QMS) requirements for medical device organizations. Typically, highly developed countries use a particular version of a standard in their regulations. A manufacturer is then required to state that they are compliant to those standards when they submit their documentation – in the form of a DoC. Drafting a European Union DoC is an important part of the CE marking process and is the final step before affixing the CE mark to your product.
Countries that Require a Declaration of Conformity
Declarations of Conformity are required in many countries, though some refer to a “Certificate” of Conformity. Countries that currently have a DoC requirement:
- Australia
- Brazil – Still in development
- China
- European Union - All countries in the EU require a DoC, but you only need to create one document
- Malaysia
- Saudi Arabia
- Singapore – Singapore will accept an EU DoC in most cases, but in certain circumstances will require a Singapore DoC
- Thailand
- United Kingdom (UK)
In the United States, the FDA’s Declaration of Conformity is built into the eSTAR program.
What is required in a Declaration of Conformity?
While the requirements vary slightly by country, a DoC should be provided on company letterhead and general requirements typically include:
- Standards related to the product that are compliant to a country’s regulations
- Product Name
- Part Number
- Physical and legal manufacturer details
- ISO Certificate Information
- Classification of the device per the country regulations and the specific rule used to classify the device
- Notified Body details
- Distributor information
- A statement regarding the regulation that the responsible party is stating compliance to
- A signature from the responsible party
Using a Regulatory Information Management (RIM) system, like Rimsys, to manage your product data and related standards will allow your regulatory team to create and control declarations of conformity and other documents. Learn more here.

An overview of 21 CFR Part 812
This document is a summary of the regulation and does include wording taken directly from the regulation itself. The original regulation should be referenced directly, however, for complete information when submitting an IDE request.
What is an investigational device exemption?
An investigational device exemption (IDE) allows a device to be used in a clinical study prior to obtaining market approval to collect safety and effectiveness data. Clinical studies are typically required to support a Premarket Authorization (PMA), but a small percentage of 510(k) applications also require clinical data when a predicate device comparison is inappropriate for the submission.
Before a clinical study is initiated, an investigational device must have an approved IDE, unless it is exempt. Devices may be exempt from IDE requirements if they are noninvasive diagnostic devices, being used for consumer preference testing unrelated to device safety or efficacy, or intended solely for veterinary use or research with laboratory animals. Refer to the full text of 21 CFR 812 for details and additional exemptions.
Part 812 – General Provisions (Subpart A)
Scope
This regulation is applicable to all clinical investigations of devices used to determine safety and effectiveness, except where exempt.
Applicability
Abbreviated Requirements
Investigations are considered automatically approved for IDEs if the device is not considered a significant risk, unless the FDA has specifically notified the sponsor otherwise. In these cases, the sponsor must still obtain IRB approval for the investigation and must comply with other requirements of Part 812, including proper labeling, record keeping, and conformed consent requirements.
Exempted investigations
IDEs are not required for devices that fall into one of the following categories:
- Devices that were in commercial distribution prior to May 28, 1976 that were used or investigated according to requirements in effect at that time. Devices that were introduced after May 28, 1976 but which have been found to be substantially equivalent to devices introduced earlier may also be exempt. This exemption is limited for Class II and III devices from the date an FDA regulation or order calls for the submission of a PMA (in the case of an unapproved Class III device) or establishes a performance standard for a Class II device.
- Diagnostic devices for which the testing is noninvasive and without significant risk, and is not used as a diagnostic procedure without confirmation through another product or procedure.
- Devices undergoing consumer preference testing, or testing of a device modification or combination of already distributed devices, if the testing is not for purposes of determining safety or efficacy and does not put subjects at risk.
- Devices solely for veterinary use.
- Devices intended for research on or with laboratory animals.
- A custom device that is not being used to determine safety or efficacy for commercial distribution.
Definitions
21 CFR Part 812.3 provides definitions for many terms, some of which are listed here.
Institution: A person, other than an individual, who engages in the conduct of research on subjects or in the delivery of medical services to individuals as a primary activity or as an adjunct to providing residential or custodial care to humans. For example, a hospital, retirement home, confinement facility, academic establishment, and device manufacturer. The term has the same meaning as “facility.”
Institutional Review Board (IRB): Any board, committee, or other group formally designated by an institution to review biomedical research involving subjects and established, operated, and functioning in conformance with part 56. The term has the same meaning as “institutional review committee.”
Investigational device: A device, including a transitional device, that is the object of an investigation.
Investigator. An individual who actually conducts a clinical investigation, i.e., under whose immediate direction the test article is administered or dispensed to, or used involving, a subject, or, in the event of an investigation conducted by a team of individuals, is the responsible leader of that team.
Monitor: When used as a noun, this term means an individual designated by a sponsor or contract research organization to oversee the progress of an investigation. The monitor may be an employee of a sponsor or a consultant to the sponsor, or an employee of or consultant to a contract research organization. Monitor, when used as a verb, means to oversee an investigation.
Significant risk device means an investigational device that:
- Is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is purported or represented to be for a use in supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a subject;
- Is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject; or
- Otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.
Sponsor: A person who initiates, but who does not actually conduct the investigation, that is, the investigational device is administered, dispensed, or used under the immediate direction of another individual. A person other than an individual that uses one or more of its own employees to conduct an investigation that it has initiated is a sponsor, not a sponsor-investigator, and the employees are investigators.
Sponsor-investigator: An individual who both initiates and actually conducts, alone or with others, an investigation, that is, under whose immediate direction the investigational device is administered, dispensed, or used. The term does not include any person other than an individual. The obligations of a sponsor-investigator under this part include those of an investigator and those of a sponsor.
Subject: A human who participates in an investigation, either as an individual on whom or on whose specimen an investigational device is used or as a control.
Labeling of investigational devices
An investigational device or its packaging requires a label with the following information:
- Name and place of business of the manufacturer, packer, or distributor (per 801.1).
- Quantity of contents.
- Statement: “CAUTION—Investigational device. Limited by Federal (or United States) law to investigational use.”
- Any additional relevant contraindications, hazards, adverse effects, interfering substances or devices, warnings, and precautions.
- If used for animal research, a statement of “CAUTION—Device for investigational use in laboratory animals or other tests that do not involve human subjects” must be on the label.
IDE Labeling cannot have any statement that is false or misleading and cannot represent the device as safe nor effective.
Prohibition of promotion and other practices
An investigational device is intended for use only within an investigation. Therefore, until a device is approved for commercial distribution by the FDA, the sponsor or investigator (or those working on their behalf) cannot:
To continue reading this Regulatory Brief, please download the full document here.

Content of FDA premarket submissions for device software functions
The Food and Drug Administration (FDA) recently released a final guidance document, “Content of Premarket Submissions for Device Software Functions.” This document is intended to provide information about the recommended documentation that medical device manufacturers should include in premarket submissions for the FDA's evaluation of safety and effectiveness of device software functions. This document replaces the FDA's “Content of Premarket Submissions for Software Contained in Medical Devices” document issued in May, 2005. Note: This new guidance does not apply to automated manufacturing and quality system software or software that is not a device.
In general, the FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidance’s describe the FDA’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited.
Highlight of changes to FDA guidance on premarket submissions for device software
Since the last time this document was revised (2005), there have been many updates to this new guidance document that are certainly worth noting. Along with the multiple changes and additions, this guide will help with more clarity in the process of determining software documentation for premarket submission and review of software medical devices, or devices that have a software function.
Some of the highlights that are important to mention include:
- Changes in terminology to more current trends. For example, the Hazard Analysis is now referred to as the Risk Management File.
- The table of contents has been extended and divided into several sections, broken down even further to include documentation type and Documentation Level.
- Level of Concern is now referred to as Documentation Levels. Although still guided by the level of product “risk," instead of major, moderate, and minor levels, they are now identified as Basic or Enhanced Levels. Basic is any premarket submission that includes device software function(s) where Enhanced Documentation does not apply. Enhanced includes device software function(s) where a failure or flaw of any device software function(s) could present a hazardous situation with a probable risk of death or serious injury, either to a patient, user of the device, or others in the environment of use.
- The Recommended Documentation section (formerly the Software-related Documentation) section has expanded to include more detail and subsections about software requirements, software architecture, software design, software development, software testing, software version history, and unresolved software anomalies.
- The Software Description has been revised to add reference to guidance about Multiple Function Device products that extends into software operation, software specifics, and software inputs/outputs.
- The Hazard Analysis is now the Risk Management file and continues to recommend ISO 14971 for guidance. It’s also been extensively revised to include the risk management plan, the risk assessment and risk management report.
- Newly added Appendix A provides many Documentation Level examples intended to demonstrate the implementation of the Documentation Level risk-based approach. Over 20+ product examples are included in this list.
- Newly added Appendix B provides examples of diagrams for the purpose of demonstrating how the System and Software Architecture Diagram could be implemented into diagrams, and to help show a clear understanding of the system and software.
In addition to this new guidance, the FDA recommends reviewing the following guidance documents (which is not an exhaustive list) for additional support in determining premarket software documentation for submission.
- Multiple Function Device Products: Policy and Considerations
- Off-The-Shelf Software Use in Medical Devices
- Design Considerations and Premarket Submission Recommendations for Interoperable Medical Devices
- General Principles of Software Validation
- Content of Premarket Submissions for Management of Cybersecurity in Medical Devices
- Cybersecurity for Networked Medical Devices Containing Off-The-Shelf (OTS) Software
- Applying Human Factors and Usability Engineering to Medical Devices

Selecting a RIM system for your medical device company
Software for medical device regulatory teams
Many regulatory affairs (RA) teams within medical device organizations are still managing their activities through spreadsheets, in-house custom-built software, or systems designed for other purposes. We believe that regulatory teams deserve purpose-built software that allows them to ensure compliance across products and markets, and provides them with the opportunity to contribute directly to revenue-driving activities.
Regulatory Information Management (RIM) solutions provide the centralized regulatory functionality needed by today’s RA teams. RIM solutions such as Rimsys are product-centric, allowing regulatory professionals to track all product-specific information and then create market submissions, link standards and essential principles, manage registrations by product by market, and control all regulatory approvals and projects. However, not all RIM solutions are created equal. If you have complex products, devices that include software, or other requirements that not all medical device companies will have, be sure to carefully evaluate potential systems for their ability to address those needs.
Justifying the need for RIM
Any software selection project begins with analyzing the need for the new software, creating a justification for the project, and obtaining the approval and budget to move forward. RIM solutions will allow your RA team to find information more quickly and operate more efficiently, which means that justification for a new RIM system typically comes from four areas:
- Cost savings – RA teams can operate more efficiently, reducing the need for outside consultants or contractors and enabling new RA team members to onboard more quickly. Better information also allows RA teams to better forecast projects in order to optimize the internal team size.
- Reduced regulatory risk – A centralized RIM system reduces multiple risks, including missing an expiration date or supplying incorrect information to a regulatory body. Even a small misstep can cause an audit finding, removal of a product from a market, or a delay in entering a new market.
- Improved competitive advantage – RIM systems significantly reduce the time that RA teams spend finding data and reacting to last-minute “emergency” requests. This advantage allows the team to drive competitive advantages and greater revenue growth through participation in market planning, product roll-out decisions, and other strategic planning. One Rimsys customer was able to improve the time for a product release by 88%. By increasing speed to market, medtech companies can recognize revenue sooner and capture more market share more quickly.
- Data harmonization – Medtech companies dealing with duplicated data and systems due to mergers or acquisitions can point to a centralized RIM system as a way to harmonize important regulatory data to ensure compliance and optimize go-to-market activities.
You will likely need to develop a comprehensive business case to support any RIM investment. This will include detailing the limitations of current approaches (including spreadsheets and costs to maintain in-house systems), quantifying the expected benefits, and explaining the evaluation process to arrive at your preferred vendor. Building this as you work through the early stages of RIM selection will prevent delays as you move into the purchase process.
Should I use external consultants to help with RIM selection?
RIM selection projects are sometimes managed by in-house teams and sometimes managed by 3rd-party consultants. How do you decide which is right for your selection project?
Does your team have experience with large system selection projects?
If your organization has a large IT team and/or digital transformation team, they likely have the responsibility of overseeing the selection of any new software systems. Be sure to understand their capabilities – have members of the team managed large system selection projects before, such as ERP or PLM selections? The regulatory team and others within the organization can provide subject matter expertise, but you will be relying on the technical team to oversee the project, define requirements, help set a budget, and more.
If the right level of expertise does not exist within your organization, an outside consultant with medical device regulatory experience and with business system selection projects should be considered. This type of consultant can be extremely helpful during the system implementation and adoption phase of the project as well.
Does our team have the bandwidth to manage a RIM selection project?
Even if your organization has an internal team with the expertise to manage a RIM selection project, they may not have the time to do so within the desired project timeframe. In this case, an external consultant can augment your existing team to get the project completed as required.
Do we need a new perspective?
Selecting a RIM solution is as much about digital transformation and process optimization as it is about ensuring you find a system with the right features. Do you have a vision of where you want the RA team to be? Have you looked at the characteristics of top-performing RA teams? If you think you might need a new perspective and an outside voice, an outside consultant may be the right choice for your project.
RIM selection project steps
Once you have determined the need for a RIM system, a selection project should include the following major steps:
Build the selection team
Put together a core selection team that consists of:
- A project leader that is typically at a manager level or above within the organization and comes from the regulatory or IT teams.
- An executive sponsor who may not participate in all aspects of the project, but who will ensure that resources are made available to the team as needed and that the project is kept on track and is aligned with overall company goals.
- IT team member(s) who will provide feedback on technical system requirements, including security and data privacy, and will support customization and integration discussions.
- Subject matter experts representing each department and team who will be using or interacting with the software.
You will add team members once you begin to implement the system, but selection teams typically consist of fewer than 10 members.
Determine requirements and selection criteria
One of the primary responsibilities of the selection team is to define the requirements for the new system, and the criteria on which systems will be judged. Requirements usually fall into multiple categories, including:
- Business requirements – These are broad requirements based on business needs. They will typically answer the question “Why do we need a RIM system?” For example, reducing the administrative burden on the regulatory team with functionality that allows the team to track registration expiration dates, create submission dossiers, and quickly report on registration status by product and market. Include project timeline and budgetary constraints here as well.
- Functional or user requirements – Functional requirements are a more detailed list of specific functions that users will need to perform in the system. For example, the ability to link standards to essential principles, manage multiple approvals for submission documents, or track UDI product data. DO include requirements that are essential for users to complete their work within your defined quality procedures. DO NOT include requirements that are unnecessarily specific (ex: the product description must contain at least 50 characters) or are so common that all systems will meet the requirement (ex: there must be a product number and description in the product record).
- System and technical requirements – Your IT team may already have a list of the overall technical requirements that all software must meet within your organization. These will likely include data security requirements and features that support system validation. Include any specific requirements regarding system availability, upgrade management, and technical support guarantees. See SaaS 101 for medtech regulatory professionals for a list of questions that you should ask a SaaS solution provider. Also include here any procurement requirements that your organization has, such as insurance requirements.
- Vendor resources and vision – You want to work with a vendor that shares your vision, not only for this system implementation but also for future growth. Evaluate each vendor’s product roadmap and plans for innovation against your organization’s digital transformation plans for the next 2-5 years. Does the vendor share your vision? Do they have the resources to support your organization and that future vision?
Establish project goals and timelines
Establish project goals and an overall project timeline. Is there a hard deadline by which the system needs to be live? What are the goals and metrics with which the success of the project will be measured? Be sure to get written agreement from the project team and executive team on the goals, timeline, and how the information will be reported.
Research RIM vendors (build your initial list)
If you are working with an experienced regulatory consultant, they may be able to get you to your short list without this step. However, if you are unsure of which systems may meet your needs, begin by:
- Researching Regulatory Information Management systems online.
- Talking to other regulatory professionals for suggestions and referrals.
- Consulting with industry analysts. Gartner's annual RIM market guide, and Gens & Associates World Class RIM report both provide an overview of RIM vendors. (Note, however, that both include both pharma and medtech-focused solutions within their respective guides.)
Build vendor short list
Based on the information gathered in the previous step, you should be able to create a short list of two to six vendors. This may require short conversations with prospective vendors, but you should have your short list before you schedule product demos and/or send out a request for proposal (RFP).
Tip: If you communicate with vendors that don’t make your short list, let them know so that they don’t continue to contact you!
Evaluate vendor capabilities
This part of the project varies greatly from company to company, but your process should ensure that all of your stated requirements are being evaluated against each vendor’s capabilities. Not all team members need to evaluate all requirements – individuals should be assigned based on their understanding of the area being evaluated. The same people, however, should evaluate the same requirements across all vendors to ensure a fair comparison.
If your organization requires an RFI (request for information) or RFP (request for proposal), those need to be compiled and sent to the vendors as the starting point for vendor evaluations. These documents allow your team to gather the same information from all vendors. Put simply, these are documents that list your requirements and ask the vendors to indicate if they address them natively within their software, through third-party integrations, or not at all. Our RIM Buyer’s Guide provides a template that can be used as a starting point.
Whatever your evaluation process looks like, your team needs to see the software. For systems as large as RIM solutions, you may need multiple demonstrations with the vendor and your team. Work with the vendor to determine how the process will work, but typically you will have an overview demonstration and then separately schedule individual sessions, if they are needed, to cover specific features or answer additional questions. While everyone on the evaluation team should attend the initial demo, additional sessions should be scheduled only when needed and only with those team members required. The following can help ensure a smooth process:
- Communicate clearly to your team what they are responsible for during the demo. Who is taking notes? Are different team members responsible for evaluating features in different areas of the software?
- Set expectations with the vendor ahead of time and maintain control of the demo agenda. The software vendor will know what sequence works best for their product, and you should allow them to guide you. However, you will want to steer them away from spending too much time on features that are not important to your team.
- Ask the vendor to keep track of unanswered questions or features that you were unable to see. The vendor should be expected to follow-up on these items.
Rate and rank vendors
Using your requirements list, each vendor should be rated for each requirement. Require vendors to clearly indicate if a requirement is met “out of the box,” requires custom development work, or is not supported at all. Consider a scale of 0-5, with 0 being a feature the vendor does not support. Multiplying the rank by the importance of the feature (3 – Critical to have, 2- Important to have, 1- Nice to have), will give you a good picture of where each vendor ranks.
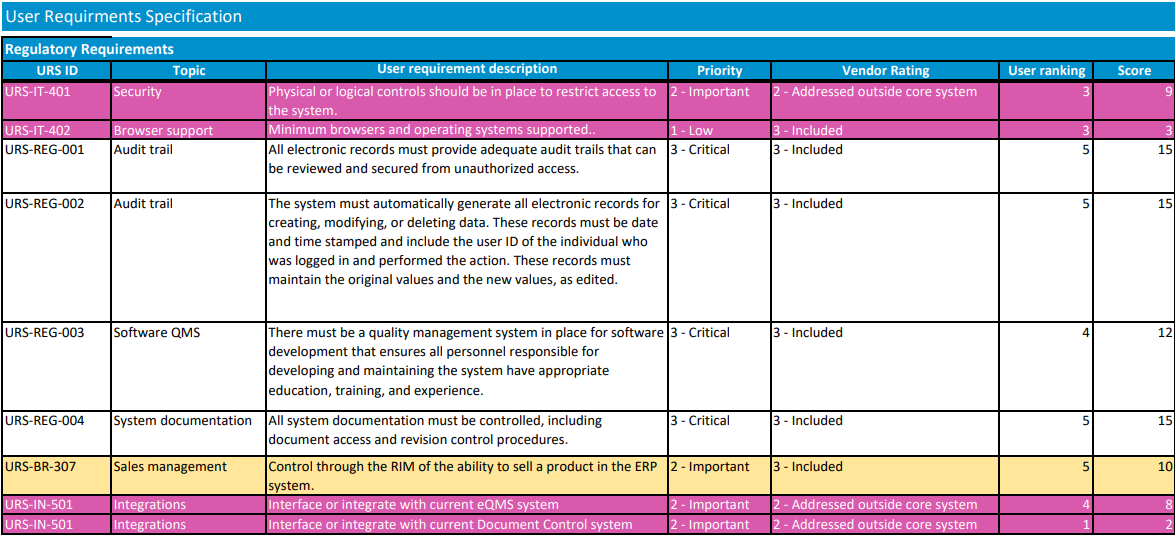
There should be some subjective items that are used in rating, also, such as how easy you believe the vendor will be to work with. Once the vendors are rated, the team should meet to discuss differences between team members' ratings and then to agree on where each vendor ranks. It is important that all requirements are considered and weighed appropriately while ranking vendors. For example, selecting the system with the best price may leave you with a vendor that doesn’t have the resources to support your implementation.
Negotiate and purchase
Hopefully, you will be able to successfully (and quickly) negotiate with your top-ranked vendor. However, it does sometimes happen that an agreement cannot be reached with the initial vendor for reasons that may include pricing adjustments or unexpected changes to the availability of their resources. In this case, you will need to move on to your second choice.
For more information on specific criteria for purchasing a RIM system, read our RIM Buyer’s Guide.
