


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.

The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR)
This article is an excerpt from The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR) ebook.
Table of contents
- Overview
- Terminology
- EU MDR/IVDR Annex I
- EU MDR/IVDR Annex II
- Proactive Monitoring & Maintenance
- Comparison Table: EU MDR/IVDR Annex I GSPRs vs EU MDD/IVDD Annex I Essential Principles
With the initial rollout of the European Medical Device Regulation (MDR) complete, medical device companies are shifting focus to the sister In Vitro Diagnostic Regulation (IVDR) which has rolling effective dates starting in May 2022. Like the MDR, the IVDR also includes new General Safety and Performance Requirements (GSPR). The expanded 2nd edition of this ebook includes a detailed summary of the IVDR GSPR regulations in addition to those of the MDR. It provides you with practical guidance on how to meet the GSPR requirements for all types of medical technology products. This ebook, however, should not take the place of reviewing the actual regulations and consulting regulatory experts when needed
Timeline
The EU MDR submission became mandatory from the previous MDD directive on May 26, 2021, and the EU IVDR effective date is quickly approaching. In fact, all submissions for new devices under the new EU IVDR must be implemented no later than May 25, 2022. Below is a high-level overview of key dates for both regulations.
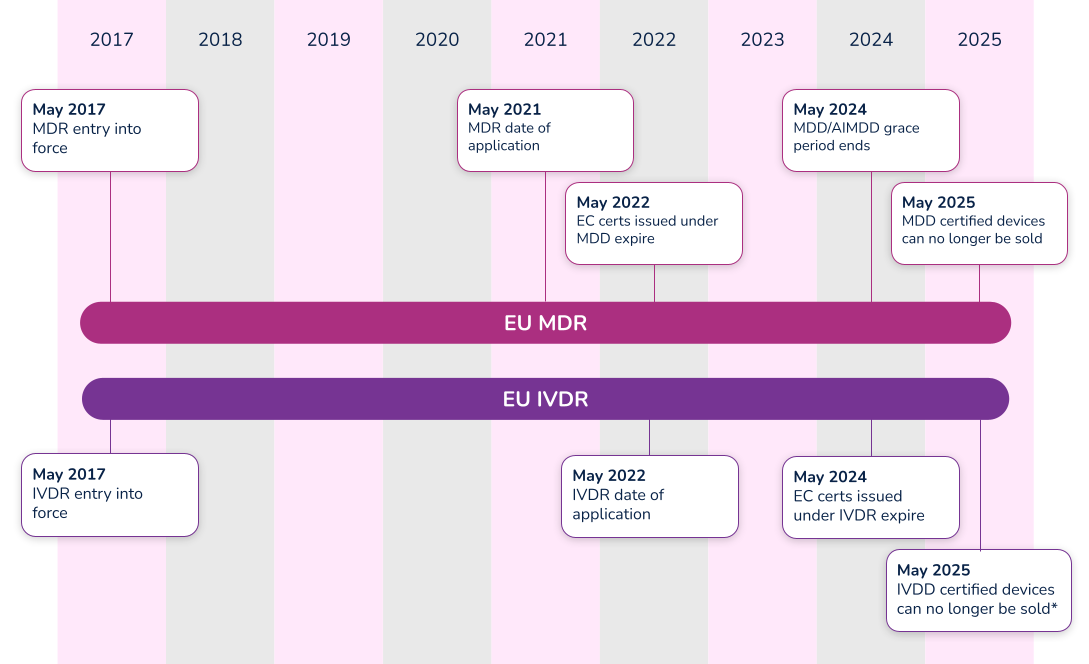
*Note that the timeline for compliance was extended in 2021. Class D (high-risk) devices have until 2025 to comply with IVDR, while Class C devices have until 2026. Class B and Class A sterile devices have until 2027 to comply with IVDR.
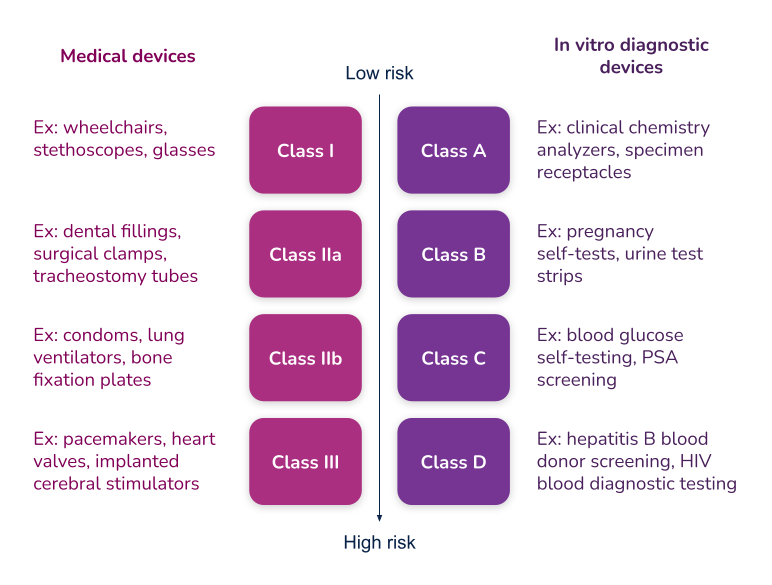
What’s the difference between Essential Requirements, General Safety and Performance Requirements (GSPR), and Essential Principles. In order to have a meaningful dialogue, let’s first discuss the three (3) main terms used in the industry.
#1 Essential requirements
The ‘Essential Requirements’ is the backbone for establishing conformity with the Medical Device Directive (MDD 93/42/EEC) and the Active Implantable Medical Device Directive (AIMDD 90/385/EEC). Detailed within Annex I of the MDD and AIMDD, the ‘Essential Requirements’ laid out the requirements that devices must meet in order to state compliance to the directives. With the implementation of the new EU Medical Device Regulation (MDR 2017/745), the ‘Essential Requirements’ will become superseded by the new EU MDR General Safety and Performance Requirements (GSPRs).
#2 Essential principles
The IMDRF laid out Essential Principles requirements in a document entitled Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices. From a high-level perspective, three basic tenets make up these ‘Essential Principles’:
- A device must be designed to be safe and perform effectively throughout its lifecycle.
- Device manufacturers must maintain all design characteristics.
- Devices must be used in a way that is consistent with how it was designed.
Many countries use the term ‘Essential Principles’ when compiling the documentation required to determine compliance to the law. For instance, the Australian Therapeutic Goods Administration (TGA) uses the term ‘Essential Principles Checklist’. Regardless of the term used, Essential Principles are of similar nature and overlap many of the Essential Requirements and new GSPRs.
#3 General safety and performance requirements (GSPR)
As of May 26, 2021, medical device manufacturers must start to comply with Annex I – General Safety and Performance Requirements (GSPRs) of the new EU Medical Device Regulation (MDR 2017/745). GSPRs are specific to the European MDR and IVDR. If you hear any other term (i.e. Essential Principles), it most likely means it is not referencing the European market.
Annex I of the EU MDR and IVDR details the specific requirements of the General Safety and Performance Requirements (GSPRs). The GSPRs are broken down into three (3) chapters in Annex I, MDR 2017/745 and IVDR 2017/746:
- Chapter 1 - General requirements
- Chapter 2 - Requirements regarding design and manufacture
- Chapter 3 - Requirements regarding the information supplied with the device
Chapter 1 - General requirements
Both the EU MDR and the EU IVDR outline General Safety and Performance Requirements (GSPRs) in great detail for medical device designers and manufacturers. The general requirements for each are almost identical and consist of the following:
- Devices must perform in a way that aligns with the intended design.
- They must not compromise the health or safety of a patient, user, or any other person associated with the device.
- Risks must be reduced as much as possible, but not so much that they negatively affect the risk-benefit ratio.
- Device manufacturers must implement and maintain a thorough, well-documented, and evaluative risk management system that continues to be updated throughout the life cycle of a device.
- Manufacturers and designers must include any necessary measures for protecting users in cases where risks cannot be completely eliminated.
- Manufacturers must provide users with information about any potential risks that remain. This information must be clear, easy to understand, and considerate of the users’ technical knowledge level, use environment, and any applicable medical conditions.
- Devices must withstand the stresses of normal use for the duration of their lifecycle. Devices must be designed, manufactured, and packaged in a way that protects them from damage during transport and storage.
- When it comes to risks and negative side effects that are known and foreseeable, designers and manufacturers must make every effort to minimize negative outcomes. They must also ensure that potential risks are acceptable when compared to the potential benefits of a device to its users.
Chapter 2 - Requirements regarding design and manufacture
The GSPRs also provide key details regarding specific information about the performance, design and manufacture of medical devices. As it relates to design inputs, the MDR and IVDR GSPRs provide highly detailed requirements relating to a device’s technical information. Further detail can be found in the comparison tables in Appendix A and Appendix B, where we have compared MDR to MDD and IVDR to IVDD.
Chapter 3 - Requirements regarding the information supplied with the device
The final key area of governance within the GSPRs relates to specific information a manufacturer must supply with a device. The general requirements for this information states that, “Each device shall be accompanied by the information needed to identify the device and its manufacturer, and by any safety and performance information relevant to the user, or any other person, as appropriate.” The requirements provide further detail as far as location - specific information that must be provided on the following:
- The device label includes its UDI.
- The user instructions.
- The packaging of a device that is intended to maintain its sterile condition.
Medical devices are subject to significant regulations and a full understanding of EU MDR and/or IVDR labeling as defined in Annex 1 Chapter 3.
In addition to the specific requirements identified within Annex I of the EU MDR and IVDR, Annex II, Technical Documentation, identifies additional requirements. Specifically, in both EU MDR and IVDR’s Section 4 – General Safety and Performance Requirements it states:
“the documentation shall contain information for the demonstration of conformity with the general safety and performance requirements set out in Annex I that are applicable to the device taking into account its intended purpose, and shall include a justification, validation and verification of the solutions adopted to meet those requirements. The demonstration of conformity shall include:
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
(c) the harmonised standards, CS or other solutions applied; and
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonised standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation.”
Let’s break this down into each part.
Requirement
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
What needs to be documented for the requirements that apply or the requirements that do not apply?
Each and every section of the EU MDR GSPR or EU IVDR should be assessed in its own right as it pertains to your medical device. When a requirement applies, a simple statement may be made that this requirement applies to the device. In practice this is often achieved using a checklist or table, with a column for applicability and a Yes/No answer against each requirement. When a requirement applies, you can move on to the other parts of demonstrating conformity regarding methods used and standards applied.
When a requirement is not applicable, a statement must be made to that effect, i.e. a ‘No’ in the applicability column. Additionally, it must be fully and properly justified. Such a justification may be something like ‘The device is not powered and is therefore not an active device. This requirement does not apply.' The justification should clearly state why the requirement has been deemed not to apply so that your notified body can understand your reasoning
Requirement
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
What is meant by “method or methods used”?
This relates to the way you complied with that GSPR requirement, historically it would be listed as a standard or other documentation reference that you have applied to demonstrate compliance, however, the question of ‘method or methods used’ is new to the MDR and it is expected that a verbal description be provided such as:
i. Risk analysis weighed against clinical evaluation benefit
ii. Performance intended demonstrated by design requirements, verification and validation
Requirement
(c) the harmonized standards, common standards (CS) or other solutions applied;
What are harmonized standards, common specifications (CS), and “other solutions”?
Harmonized standards
These are standards that have been specifically developed and assessed for compliance to a regulation or directive. They are published in the Official Journal of the European Union (sometimes just referred to as ‘the OJ’) and if you comply with these standards then there is a ‘presumption of conformity’ with that directive or regulation to which they have been harmonized. These harmonized standards can only be created by a recognized European Standard Organization (such as CEN or CENELEC). When a standard is harmonized, an annex is added that describes how the standard conforms to the directive or regulation. When using harmonized standards, you should make sure that you understand how the standard conforms so that you do not claim compliance when the standard either does not meet that requirement or only partially meets that requirement.
If a standard does not meet a certain requirement of the directive or regulation, or indeed only partially meets it, then you must employ additional mechanisms for compliance. If a harmonized standard meets part of a directive or regulation, then by complying with that standard you also fully meet the corresponding requirement(s) The list of harmonized standards continues to grow - refer to the “Healthcare Engineering” section of the European Commission’s Harmonized Standards page for current information. In this case, using an MDD harmonized standard and documenting a justification for doing so (i.e. how you believe the standard demonstrates compliance with the GSPRs), should provide sufficient evidence
Common specifications
Common Specifications (CS) are a new concept in the MDR. They allow the European Union to add additional requirements that must be met in order to claim compliance where harmonized standards do not exist or where relevant standards are considered insufficient. The definition of a Common Specification is:
‘A set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.’
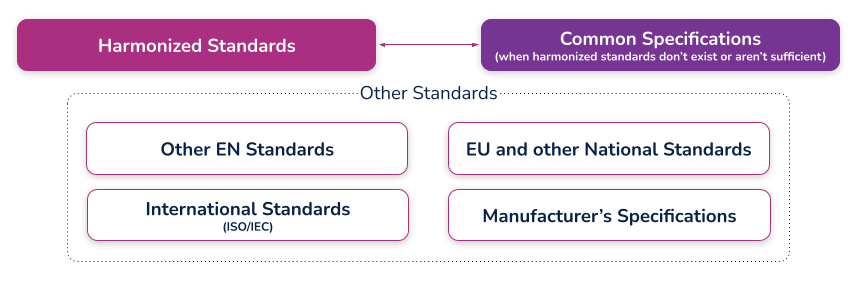
Requirement
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonized standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross- reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation;
What is the expectation for incorporating a "cross-reference to the location of such evidence within the full technical documentation"?
This means that someone looking at the document should be able to identify exactly where in the technical documentation that the compliance evidence can be found. For example, this may refer to test reports and their exact location, or it could even reference locations within a large document, depending on the GSPR and your particular documentation. (i.e. if you have included usability risks as part of a larger risk assessment, you may need to say ‘See Technical File XXX, Section XX, Doc RMF001 rev 3 lines 65-78’). In other cases it could just mean the whole document reference, i.e. Have you done risk management? – then yes, it is RMF001 rev 3. What the specific reference actually is depends on how you have managed your technical documentation and how defined it is (i.e. separate reports or one big one). There should be no ambiguity as to where the document is located
An example of a completed GSPR checklist could look something like this (applicable and nonapplicable examples are shown):
Specification developers and manufacturers must continually maintain their technical documentation to stay compliant. Part of this process is to ensure that they take into account the "generally acknowledged state of the art".
Proactive monitoring
'State of the art'
There is no formal definition of ‘state of the art’ within the EU MDR or IVDR, although it is mentioned many times. ‘State of the art’ is an ongoing debate; however, it generally means that it embodies what is currently and generally accepted as good practice in the medtech industry. The ‘state of the art’ does not necessarily imply the most technologically advanced solution.
One consensus on state of the art is being up to date and compliant with the current and in effect standards that are applicable to your device. This means that if a standard is updated that your medical device is compliant with, you must evaluate that update to ensure that it would meet the EU MDR or EU IVDR ‘state of the art’ requirement. This is not a new requirement from the EU MDD but it is spelled out more clearly in the EU MDR.
The specification developer or manufacturer is ultimately responsible for determining if the updated standard applies or does not apply to their device(s). Either way, the justification should be documented within a gap analysis.
Monitoring for changes
Of course, 'state of the art' only applies if you actually know if something changed. This is why you need to develop a process for monitoring the standards that compliance is claimed. Every single standard that is associated with your technical documentation must be actively monitored, reviewed, and reported on.
If you have a product on the market and need a better way to monitor and maintain your General Safety and Performance Requirements (GSPR) or Essential Principles, Rimsys can help. Rimsys digitizes and automates GSPR and Essential Requirements so you can dynamically update and proactively monitor changing standards and evidence files.
When a standard or evidence file changes, you will automatically be notified and can update one GSPR or all of your GSPRs as applicable with a single click of a button. If additional information is needed, such as testing, it’s also invaluable to ensure that all devices are identified. What used to take weeks of manual, error-prone administrative tasks is now done in seconds within a fully validated, secure, maintenance-free, cloud-based solution
Maintenance
Maintaining and updating your technical documentation is generally the hardest part of staying compliant. Robust processes must be established to ensure nothing slips through the cracks and show up as nonconformances during regulatory audits.
Gap analysis
In addition to meeting the ‘state of the art’ requirements and the continuous proactive monitoring of standards, once a change has been detected that affects the technical documentation, a proper and thorough gap analysis must be completed.
The gap analysis between the old versions and the new versions, or an evaluation of a brand new standard, must occur and be properly documented. The gap analysis should detail what is applicable and what is not applicable, with your supporting justification.
If something within the new or revised standard was applicable to your device, additional engineering testing, documentation, justification, and, in some instances design changes, may be needed to ensure compliance
GSPR updates
Once the gap analysis has been properly documented, specification developers and manufacturers must update their GSPRs.
These updates include finding the withdrawn or superseded standard or evidence file throughout each row within your GSPR table, for every single device on the market on which this change is applicable. This could be one table or dozens of tables depending on the complexity of the products and your product mix.
Without a holistic RIM system to help you, this is an error-prone process as is it tedious, administrative, and extremely easy to miss an inappropriate referenced standard or evidence file.
Extreme diligence on the regulatory or engineering team must occur to ensure these critical updates to the GSPRs are not missed and a gap analysis must be properly referenced throughout. Any justification for including or excluding a new standard or evidence file will be scrutinized by regulatory auditors, and without proper maintenance, may lead to additional review time.
To continue reading this eBook including Comparison Table of the EU MDR Annex I GSPR vs. the EU MDD Annex I Essential Requirements, please register to download the full version.

The beginner's guide to the FDA PMA submission process
This article is an excerpt from The beginner's guide to the FDA PMA submission process ebook.
Table of Contents
- Introduction
- PMA basics
- FDA interactions
- Contents of a traditional PMA submission
- PMA supplements and amendments
- PMA Quality Management System (QMS)
- Review process and timeline
If your organization is planning to market a new medical device in the United States, you first need to determine which regulatory class the device falls under. The vast majority of medical devices regulated by the FDA are either Class I or Class II medical devices, requiring a 510(k) premarket notification or a simple registration if exempt from 510(k) requirements. However, if your device sustains or supports life, is implanted, or presents a “potential unreasonable risk of illness or injury,” your device is likely a Class III device which will require Premarket Approval (PMA) from the FDA before it can be marketed in the United States. Novel devices, for which there are no existing substantially equivalent devices, are automatically classified as Class III as well. Novel devices with a lower risk profile, however, may qualify for the De Novo process instead of the PMA. Just 10% of devices regulated by the FDA are Class III devices.
This ebook provides an overview of the PMA process and its requirements, but it is not designed to be the only resource used in compiling a PMA submission. The FDA provides significant documentation on this process, starting with the regulation governing premarket approval that is located in Title 21 Code of Federal Regulations (CFR) Part 814.
FDA: Background and device oversight
Before we explain what a PMA is, let’s first talk generally about the Food and Drug Administration (FDA) and device oversight. The FDA is the U.S. governmental agency responsible for overseeing medical devices, drugs, food, and tobacco products. When it comes to medical devices, the FDA’s mission is to “protect the public health by ensuring the safety, efficacy, and security of...medical devices.” At the same time, the FDA also has an interest in “advancing public health by helping to speed innovations.” In other words, the FDA’s goal is to make sure devices are safe and effective for public use, while also ensuring that devices have a quick and efficient path to market.
In order to achieve this balance of safety and efficiency, the FDA has three different levels of oversight depending on the risk level of the device: (1) exempt from premarket notification, (2) Premarket Notification, also known as 510(k), and (3) Premarket Approval (PMA).
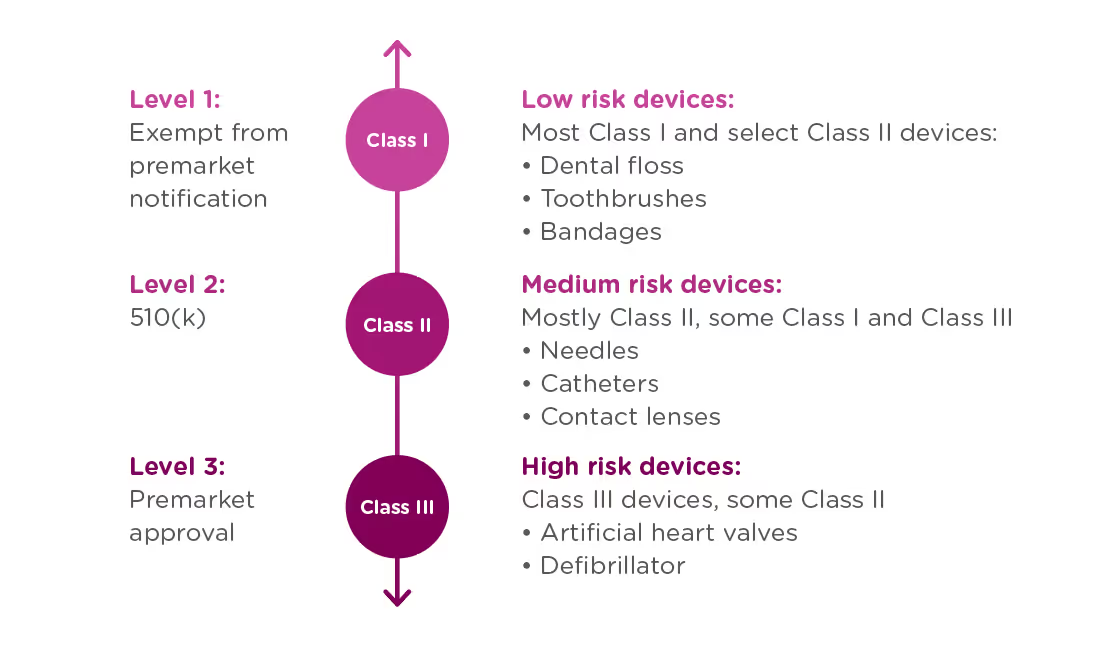
When is a PMA required?
The PMA process is the most stringent regulatory process for medical device approval under the FDA and applies to almost all Class III devices. To determine whether your device requires a PMA, you must first Classify your device by searching the Product Classification Database. The database will provide you with similar devices; their name, classification, and link to the Code of Federal Regulations (CFR) if applicable.
- If a substantial equivalent is found in the Product Classification Database with a submission type of 510(k), you should submit a 510(k), not a PMA.
- If the product classification database identifies your device as Class III and/or requiring a PMA - you should submit a PMA.
- If your device involves a new concept and does not have a classification regulation in the CFR, the database will list only the device type name and product code. In this case, the three-letter product code can be used to search the PMA database and the 510(k).
- If your device cannot be found in the product classification database because it is a new type of device and should be classified as a Class III device because of the level of risk it presents*.
Class III devices support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential and unreasonable risk of illness or injury.
Note that if your device is a new concept without a substantial equivalent, but does not present the level of risk of a class III device, it may be eligible for the De Novo process as a class I or class II device.
PMA vs 510(k)
Not only are PMA and 510(k) processes applicable to different types of devices, they have different purposes.
510(k): A 510(k) is intended to demonstrate that the device for which approval is being sought is as safe and effective as a currently marketed device that does not require a PMA.
PMA: A PMA is intended to prove that a new device is safe and effective for the end user. A PMA is much more detailed and in-depth than a 510(k). Device manufacturers are typically required to present human clinical trial data, in addition to laboratory testing data.
The difference in complexity between a PMA and 510(k) also affects the time needed to process the submissions. The FDA typically accepts or rejects a 510(k) submission within 30-90 days, at which point the device is posted to the FDA’s 510(k) database. A PMA submission can take up to 180 days to be processed, at which point the FDA can approve or deny the application. The FDA may also issue an “approvable” or “not approvable” letter, which the applicant can choose to respond to, thereby adding time to the submission process.
PMA application methods
There are a number of types of PMA application methods. While most devices which require a PMA will follow the traditional process, be sure to verify that you are using the correct application process to maximize your chances for success and avoid unnecessary delays:
Traditional PMA
The most common method for attaining FDA clearance for Class III devices, the traditional PMA is the appropriate option for most devices that have completed clinical testing.
Modular PMA
The modular PMA is the appropriate application method for devices that have not yet completed clinical testing. Applicants complete individual “modules,” with final confirmation granted once all sections are completed. For additional information on specific requirements of a modular PMA, read the FDA’s Premarket Approval Application Modular Review.
Product Development Protocol
Use the Product Development Protocol (PDP) with medical devices that are based on well-established technology. The PDP process for gaining market approval merges the clinical evaluation and development of information, and involves an agreement between the manufacturer and the FDA. The process provides the advantage of early predictability for the manufacturer and allows early interaction that can identifyFDA concerns as soon as possible in the development process. Because the PDP identifies the agreed upon design and development details, a completed PDP is considered to have an approved PMA. For additional information, read more about the FDA’s PMA Application Methods.
Humanitarian Device Exemption
A Humanitarian Use Device (HUD) is specifically defined as a device intended to benefit patients that are affected by a disease or condition that affects less than 8,000 individuals in the U.S. per year. TheHumanitarian Device Exemption (HDE) approval process is designed to encourage clinical activity around rare conditions, and does have certain restrictions, including:
- After receiving HDE approval, a HUD is eligible to be sold for profit only if the device is intended to address a disease or condition that occurs primarily in pediatric patients, or occurs in pediatric patients in small numbers.
- If an HDE is approved to be sold for profit, the FDA will determine an annual distribution number(ADN). Any devices sold beyond the ADN limit are required to be sold for no profit.
For more information see the FDA’s explanation of the Humanitarian Device Exemption.
CBER Submissions
There are two centers within the FDA responsible for evaluating medical devices. While the majority of devices will go through the Center for Devices and Radiological Health (CDRH), some will be managed by The Center for Biologics Evaluation and Research (CBER). CBER regulates medical devices related to blood and cellular products, including blood collection and processing procedures as well as cellular therapies. This ebook focuses on submissions made through the CDRH, but you can view CBER Regulatory Submissions – Electronic and Paper for more information on the CBER process.
To continue reading this eBook, including a walk through of the different types of required and optional FDA meetings and communications, a detailed list of the contents of a traditional PMA submission, and an overview of quality management system requirements, please register to download the full version.

An overview of 21 CFR Part 11 regulations for medical device companies
What is 21 CFR Part 11?
21 CFR Part 11 refers to the federal regulation that address electronic records and electronic signatures associated with FDA requirements. This single, relatively small, part of the Code of Federal Regulations is extremely significant for companies with FDA-regulated products because it impacts every document signature, electronic file, and FDA submission. Codified in 1997, interpretations of this FDA-issued regulation continue to be debated and re-evaluated as the technology supporting electronic records and signatures changes. In this article, we’ll discuss the regulation and generally accepted interpretations.
Note that discussions and statements in this document are our observations only and should not be taken as fact. You can refer directly to the regulation here.
Part 11: General Provisions
The General Provisions section of 21CFR11 addresses the scope of the regulation, when and how it should be implemented, and defines some of the key terms used. It states that the purpose of Part 11 is to define the criteria under which electronic records, electronic signatures, and handwritten signatures attached to electronic records are equivalent to, and as reliable as, handwritten signatures on paper documents.
Fundamentally, any record that is maintained, used, or submitted under any FDA records regulation is subject to Part 11, and the FDA will accept electronic records in lieu of paper records if an organization can prove that their records and systems meet the Part 11 requirements.
The General Provisions subpart also sets forth a number of definitions, and we’ve listed the ones that are most significant to our discussion here:
- Closed System: A computer system or software whose access is controlled by the same people who are responsible for the information stored in the system. Because the opposite of a closed system, and “open system,” is subject to additional scrutiny be sure that you are able to thoroughly explain and provide documentation for a decision to classify your system as a “closed system.”
- Open System: A computer system or software whose access is not controlled by the same people who are responsible for the information stored in the system.
- Digital Signature: An electronic signature created in a manner that can be verified, ensures the identity of the signer, and maintains the integrity of the document and signature. This often involves the use of cryptography and/or biometric data.
- Electronic Signature: Symbols that represent a legally binding equivalent to an individual’s handwritten signature (as adopted and authorized by the signer).
Part 11: Electronic Records
The Electronic Records section sets forth the requirements for administration of closed and open electronic record-keeping systems, then discusses signature manifestations and requirements for establishing a link between signatures and records.
Part 11 defines a “closed system” as any computer system in which the users controlling access to the system are the same people who are responsible for the data in the system. Today, most systems can be classified as closed systems, but take special care to document control procedures around software that is hosted offsite or classified as a SaaS solution.
This section of the regulation deals with the controls that need to be in place for all applicable electronic record systems by defining:
- Procedures to ensure that all electronic records are authentic, have integrity, and can ensure confidentiality (where that is appropriate).
- Validation requirements for systems that maintain electronic records to ensure that all records are accurate, reliable, and that the system performs consistently according to regulatory requirements.
- Audit trail requirements for all regulated records to ensure a complete history of all changes to records are maintained.
- Controls around system access and document signatures.
Part 11: Electronic Signatures
The Electronic Signatures section defines the components of electronic signatures and the required controls and procedures necessary for using them.
In general, an organization must be able to demonstrate that electronic signatures:
- Are unique to each individual, and that the individual assigned an electronic signature has had their identity and level of authorization verified.
- Must be based either on biometric data (such as fingerprints) or made up of two distinct pieces (ie: a User ID and password)
- Require appropriate controls to ensure that they are verified periodically, cannot be used by someone other than the intended user, and are immediately deactivated if compromised in any way.
Practical application of 21CFR Part 11 for regulatory affairs professionals
21 CFR Part 11 is a critical regulation, and one that can be open to interpretation. Below, we cover some of the key areas that should be of concern for RA professionals. This is an overview of key areas only, and should not be taken as complete instruction or guidance for 21CFR part 11 compliance.
System compliance and validation
Any system that you are using to store electronic records that fall under FDA regulations needs to be compliant with Part 11. This includes everything from spreadsheets to full-featured RIM and document management systems.
Software vendors will often document how their systems are developed to be compliant, and may even support system validation during implementation - but it is ultimately the responsibility of the user organization to ensure that their systems and processes are compliant with Part 11. System validation is the process of documenting that your system meets all of the Part 11 requirements. Software vendors can support this process by ensuring that their systems are built on a highly secured infrastructure that can be demonstrated and proven.
The Rimsys system was built from the ground up to meet the stringent requirements of not only 21 CFR Part 11, but other industry standards and good practices guidelines (GxP). We have put in place a rigorous validation program, built by industry experts and supported by a secure and well-documented infrastructure. For more information, visit the Rimsys Security and Privacy page.
Audit trails
Audit trails are the required system logs that track the who, when, and what of every change made to data that falls under Part 11. Audit trails should be generated and time-stamped by the system, with no ability for users to change that information. Audit trails serve two purposes under 21 CFR Part 11:
- To demonstrate that documented policies and procedures are being followed, including that only users with the appropriate authority are managing data.
- To prove that data retention policies are being adhered to (see below).
At any time, you should be able to view the history of any record, from a Design History File to a submission document, in order to determine what changes have been made, when they were made, and by whom.
Record retention
21 CFR Part 11 specifies that electronic records must be protected and readily available throughout the defined record retention period. Additionally, 21 CFR Part 820 specifies that records related to the quality, manufacturer, regulatory submissions, or any other data that falls under FDA regulation, should be maintained for the life of the medical device and for a minimum of two years from the date of first commercial distribution. This is often referred to as “cradle to grave” tracking.
This means that regulatory professionals need to not only be aware of their company’s record retention policy, but need to ensure that any system being used to track regulatory submissions or other data subject to audit meets Part 11 and Part 820 requirements. Note that record retention requirements apply also to paper records where they are the source document.
Electronic and digital signatures
An important piece of 21 CFR Part 11 is its definition of electronic and digital signatures. “Electronic signature” is used to define any set of symbols that are used in place of a handwritten signature, whereas a “digital signature” is an electronic signature based on methods that ensure the identity of the signer where the integrity of the data can be verified. A digital signature can be based on biometric data (such as fingerprints) or secure user IDs and passwords that are controlled to ensure only one authorized user can use the signature.
As a regulatory affairs professional, you should ensure that:
- Everyone on your team who needs to sign documents has their own unique digital signature and understands the importance of protecting it. Sharing of electronic credentials is a common FDA audit observation. Also ensure that users who are not required to sign documents have appropriate access to data to discourage other users from sharing login credentials with them.
- You are following your company’s policies concerning electronic signature audits so that passwords remain updated and strong and signatures are revoked when a user leaves or changes positions.
- You immediately report any possible loss, theft, or sharing of user credentials or devices that generate identification codes.
While 21 CFR Part 11 is usually considered more of a “quality regulation,” it is important that regulatory teams within medical device organizations fully understand this regulation and its compliance implications. To learn more about the regulations, click below to read our regulatory brief.




Announcing the Rimsys advisory board
Rimsys Regulatory Management Software, the leading Regulatory Information Management (RIM) platform designed specifically for the medical device industry, is proud to announce the creation of its prestigious advisory board.
By creating an advisory board with the most forward-thinking minds and preeminent talent in the medical device industry, Rimsys is now aligned and positioned to continue its growth and mission as the leading regulatory management software in the medical device industry.
The board members serve as strategic partners in the continued development and success of Rimsys, as a catalyst to achieving its short-term and long-term goals. The board is comprised of trusted thought leaders, known for being change agents in the industry and having the respect of their peers throughout their career and community.
"The management team could not be more pleased with the addition of these board members and involvement they have with the direction of our company. We are fortunate and thrilled to have such talented and experienced industry veterans and look forward to their many contributions,” said James Gianoutsos, Founder & President of Rimsys."
As advocates and ambassadors of Rimsys, the board supports the management team through strategic analysis, consultation, and providing professional expertise and guidance to help navigate and mitigate potential risks, discover opportunities, and define benchmarks for continued success and organizational growth.
"Rimsys is a very unique product in the marketplace, so it’s only fitting that we bring on such unique minds to the board. Their expertise and vision are exactly what is needed to help us improve our business, our technology and expand our product offerings,” said Brad Ryba, CTO of Rimsys."
The current advisory board members include:

John Speer
Jon Speer has over 20 years of experience in the medical device industry that includes quality management, product development, and project management at Creo Quality, Cook Inc., Theron Inc., and Maetrics LLC. Jon is experienced in managing multiple projects and taking medical device concepts through development, regulatory submission, and ultimately to market. Additionally, Jon is an expert in the design and implementation of FDA-compliant quality management systems and is an active contributor at MedCity News, Med Device Online, Quality Digest, QMed, and is the host of the #1 most downloaded podcast in the industry, The Global Medical Device Podcast. Jon currently serves as the Founder and VP QA/RA at Greenlight Guru, an eQMS that is specifically designed for the medical device industry.

Chris Ferguson
Chris Ferguson has over 15 years of global medical device quality and regulatory affairs experience managing class I, II, and III medical devices and consumer products for numerous world-class global organizations. Chris has successfully led global quality and regulatory projects and teams through FDA, ISO, consumer safety audits, and quality system remediation activities and has in-depth knowledge of the current regulatory landscape. Chris currently serves as Director of Quality Assurance for TransEnterix, Inc.

Bruce McKean
Bruce McKean has over 25 years of medical device industry experience as a regulatory professional specializing in quality and regulatory (Q&R) compliance, design controls, and Q&R related mergers and acquisitions. During his career, Bruce has focused on implementing and maintaining design controls, product submissions, quality management systems internal to his company and for newly acquired companies, corporate Q&R internal audit program, and performing Q&R due diligence audits on target companies. Most recently, Bruce has led a corporate-wide MDSAP compliance initiative and is focusing on the EU MDR implementation. Bruce currently serves as Director of Q&R Operations at Philips Healthcare.

Adam Price
Adam Price has over 15 years of medical device industry experience as a quality assurance and regulatory affairs professional. Adam is currently focused on the development of strategies and solutions to establish and maintain compliance in today’s fast-paced regulatory environment to enable businesses to meet the demands of the global market. Adam is cognizant of dynamic and complex market requirements and the need for effective tools and solutions to allow businesses to maintain continued regulatory compliance. Adam currently serves as Head of Post-market Surveillance at Philips Healthcare.

The 510(k) application: if content is king, then communication is queen
Often, the first thing we hear from a consultant or a medical device company regarding an FDA 510(k) premarket notification is that it was delayed because the FDA reviewer did not understand something simple within the application, or completely missed it.
What is wrong with the reviewer? How could they have missed something so simple? I couldn’t have been any clearer!
Sound familiar?
FDA is overworked, under-resourced, and will most likely miss something simple in your file upon reviewing.
As the specification developer, you know the design and history of the product better than anyone. You are providing that entire history in a formal application for review, and hopefully, clearance. A basic understanding of the technology is a must; however, think about the situation from the FDA reviewers’ point of view. 510(k) applications are inherently technical and sometimes need a brief discussion with the FDA reviewer for clarification or a general overview of your device.
Starting this dialog earlier is important for a smooth path to clearance. Part of this process involves requesting a Pre-Submission (“Pre-Sub”). Pre-Subs are a type of feedback that is part of FDA’s Q-Submission program.
Pre-Subs
Pre-Subs are a formal written request from an applicant for feedback from FDA to be provided in the form of a formal written response or a meeting (in-person or teleconference) in which the feedback is documented in meeting minutes. A Pre-Sub provides the opportunity for a submitter to obtain FDA feedback prior to intended submission of a premarket submission (i.e., IDE, PMA, HDE, De Novo request, 510(k), Dual, BLA, IND) or Accessory Classification Request, among others.
Pre-Subs are entirely voluntary on the part of the applicant. However, early interaction with FDA and careful consideration of FDA’s feedback may improve the quality of subsequent submissions, shorten total review times, and facilitate the development process for new devices.
Pre-Subs provide FDA reviewers with an introduction to you and your device rather than just having a 510(k) application dumped on their desk. FDA reviewers appreciate Pre-subs because they can get a sense of when they should anticipate filings and can plan their workloads accordingly.
FDA reviewers, like all of us, only have a certain amount of time during the day. If they are unable to find information easily or do not properly understand something, then they may state that the relevant information is missing from the application or needs further clarification. This kicks the 510(k) application back to you and stops the review clock. That is directly on the industry submitter, not the FDA reviewer.
The bottom line
FDA reviewers are people too. This is an obvious but often overlooked point to make. Sometimes they miss simple (and sometimes seemingly apparent) information. They make mistakes. The last thing you want to do is start yelling or pointing fingers. After all, you don’t want to burn any bridges as you will most likely deal with the same FDA reviewer upon subsequent submissions for similar products. Always be timely, concise, straightforward and respectful.
At the end of the day, keep in mind that your FDA reviewer isn’t as familiar with your medical device as you are. You need to help them understand items that are unclear, and the only way to do that is through building the communication channel early and having constructive conversations.
Did you know Rimsys Regulatory Management Software will keep track of all communications, notes, decisions, and tasks associated with your 510(k) application and other international regulatory submissions? Find out more now with a free demo and we will show you the power of the only regulatory information management (RIM) system platform designed specifically for the medical device industry.

Introducing government submission templates (i.e. 510k, STED, CSDT) and more!
Rimsys released a major revision on Dec. 3, 2018 that included adding registration workflows, registration owners, Kanban boards, new registration dates (e.g. anticipated approvals dates) and registration lifecycle stages.
Rimsys has been working aggressively over the last month to finish up the final touches on our next release, and we are excited to tell you that it has been officially released! These new features will benefit any size of an organization and continues our pathway to better serving the regulatory affairs professionals in the medical device industry.
Here are a few of our features released:
- Document templates – Depending on where you are registering your product, you can now choose or create your own document template that your team can follow to keep you compliant, better organized, and standardize your regulatory process. A few of our templates include: Summary of Technical Documentation (STED) for IVD and non-IVD Medical Devices, ASEAN Common Submission Dossier Template (CSDT), 510k Template, and more!
- Multi-product registrations – You can choose 1 or 1000 products (at the part number level) to register simultaneously into one market.
- Bulk search & replace for essential principles – We have been working with a few of our customers to get this functionality rolled out by the beginning of January. You now have the ability to search / replace / or remove a standard or a document throughout multiple essential principle tables simultaneously. Let’s say you are managing 10 (or even 500) essential requirements checklists…with a few clicks of a button, you can search, find and replace 1 (or all) standards or documents in EVERY table! If you have ever managed an essential requirements checklist before, we can’t stress enough of how HUGE of a time saver this is for you and your team!
- Embedded documents in essential principles – We now automatically embed your objective evidence directly into the Essential Principles PDF record. This means that when you export your essential principles as a PDF, every single document that is linked to it will be embedded directly into the searchable PDF. You never have to go looking for documents again!
- Dashboard updates – Added key metrics so your team can all be on the same page
- Expanded reporting capabilities – Added the ability to drill-down into key metrics
With this release, Rimsys will be better positioned to cater to organizations of all sizes. We have even more features and modules coming out in the coming months that will further enhance the benefit you receive from using Rimsys.
What’s next?
Rimsys has been working hard to be the single source of truth of all things regulatory related for medical devices. One of the most frequently requested features from our customers is the ability to bring regulatory updates on regulations, laws and guidance documents directly into Rimsys. We are happy to report that this feature has been in development for quite some time and we will be releasing in the next couple of months.

MDSAP device marketing authorization and facility registration
What is the medical device single audit program (MDSAP)?
The International Medical Device Regulators Forum (IMDRF) recognized that a global approach to auditing and monitoring the manufacturing of medical devices could improve their safety and oversight on an international scale. This created the Medical Device Single Audit Program (MDSAP) and allows a recognized Auditing Organization to conduct a single regulatory audit of a medical device manufacturer that satisfies the relevant requirements of the regulatory authorities participating in the program.
To date, the MDSAP participating countries include:
- Australia (Therapeutic Goods Administration – TGA)
- Bazil (Agência Nacional de Vigilância Sanitária)
- Canada (Health Canada)
- Japan (Japanese Pharmaceuticals and Medical Devices Agency)
- United States (FDA)
The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Programme and the European Union (EU) are Official Observers, which means they are waiting for the results of the pilot MDSAP program to determine if it’s worth their while to sign on as an official partner.
When does MDSAP come into effect?
Starting January 1, 2019, if you’re selling medical devices into Canada, it’s not optional and you must be certified to MDSAP, or at the very least, show evidence that you are in the process of complying.
As part of the MDSAP auditing program, there are seven chapters an auditor must cover. One of those chapters is specific to marketing authorization and facility registration, which also touches on two other chapters, management and design development. An auditor will be specifically looking for the following:
- Have you complied with requirements to register and/or license your device facility;
- Did you submit device listing information;
- Did you obtain device marketing authorization;
- Have you arranged for assessment of changes and obtained marketing authorization for changes to devices or the quality management system which require an amendment to existing marketing authorization
You must have that information organized in a meaningful way that you can get to it quickly and show, objectively, that you fulfilled the requirements of MDSAP and all of the country regulatory requirements that fall under MDSAP. That also goes hand-in-hand with ISO 13485:2016 where you need a controlled release of products into the appropriate jurisdiction. If you’re trying to be a global leader or a global company, for that matter, in this day and age, you need to have a solid system in place to manage those marketing authorizations worldwide.
Controlled release of product
If you are selling out of the United States, you must comply with the laws of each importing country. That simply means, no matter where you sell outside of the United States, you must meet the importing country’s requirements for marketing authorization. Your regulatory team and business need to be on point by having a robust regulatory system in place that upon product release, you’re meeting those specific requirements. You must have a mechanism in place to ensure that you don’t release product prior to it being properly registered.
That mechanism starts during product realization. Sales, marketing, customer service, engineering, operations, and regulatory teams must all be on the same page. Often times, regulatory is perceived as the bottleneck to product release. However, this is a misconception and is primarily driven by poor planning during the design and development process.
Auditing to MDSAP
Auditors are looking for the standardized process for controlling the release of the product and ensuring that the process has been adequately established and implemented within your facility. MDSAP has a very rigid auditing process to ensure the proper market authorizations have been obtained and facility registrations have occurred.
When your company is audited, an auditor will request records from product outside of the MDSAP participating countries due to the broad jurisdiction of US and international regulations. If the auditor finds issues with those products, they can draw that parallel to determine that your company doesn’t have a controlled product release process and you need to investigate to ensure there isn’t a systemic issue. That means an audit observation and a corrective and preventive action (CAPA) plan need to be established to rectify the issue(s).
What does this mean for medical device manufacturers?
A regulatory professional’s job is worldwide nowadays, which means it is a lot of responsibility, burden and business risk that are on their shoulders. Do you really want all of that being managed by excel files, outlook reminders, and disjointed processes? It must be a fundamental, standardized process, ingrained into your quality management system, that you need in place in order to NOT run into any compliance issues. Your organization must have a standardized process to ensure that your company is releasing good (and approved) product into the market while maintaining any changes to that product (and registration) while it’s in that market.
The requirement is not only that you get the marketing authorization, but you stay compliant when you’re already in that market. That means you must constantly be monitoring for expiring registrations, any type of design changes with your product, and how they affect your marketing authorizations within those countries.
From a quality management system standpoint, you need a good change control process in place that ties directly to your regulatory team. If you don’t have a good regulatory process now, you’re not going to have one later. It’s going to be too late, and the amount of information that your regulatory team must handle today is only going to increase. That’s why you must develop those systems now.
To learn more about the MDSAP, markets where it’s applicable, pros and cons of using MDSAP vs Regulatory Authority inspections, and audit sequence and grading, download our Ultimate Guide to MDSAP.

Top 6 benefits of a regulatory information management (RIM) system for medical devices
The medical device and in-vitro diagnostic medical device industry are in dire need of a robust, practical and easy to use regulatory information management (RIM) system. Without a unified and collaborative system, serious consequences to your business can occur, including an increased risk of non-compliance, increased costs as well as a possible significant reduction in a product’s revenue potential.
1. Revenue impact
Missing registration dates, slow-to-market losses, and long-term, cascading impacts such as loss of customer loyalty have an immediate impact to market capitalization. Moreover, improper release of product due to lack of visibility to regulatory statuses can cause fines and loss of credibility with authorities, which can result in increased scrutiny.
2. Regulatory compliance
Compliant product releases are required in the medical device industry. Automation that creates safeguards to prevent unintentional release of products into markets is a must. Regulators from different markets are working together to identify instances of non-compliance as well as misalignment of information in submissions and other communications. More effective control of the submission, enabled by a unified platform, can lead to a leaner, higher quality submission and a reduced regulatory burden.
3. Faster time to market
Better planning and tracking in a unified system can monitor process metrics, milestones, and automatically informing submissions plan timelines with actual performance. A unified solution connects planning to execution, allowing improved, real-time process monitoring. Teams can quickly spot constraints and take action, allowing the product to get through your process faster.
4. Efficiency and collaboration
Regulatory processes touch multiple functional areas. Regulatory functions have been piecing together disparate systems to achieve marginal improvement. This landscape inhibits the accurate and timely transfer of data and disruption in cross-functional workflow.
5. Efficiency and collaboration
Employee turnover on regulatory teams is linked to the stress and increases greatly if team members consider processes to be inefficient or wasteful. Being able to perform one’s job efficiently and the perception of being part of a high-performing organization contributes to employee satisfaction and retention.
6. Insurance policy
Having a fail-safe in place now for when (not if) your top talent leaves prevent the loss of company and product specific tribal knowledge. Retraining a new employee without the subject matter expert can cause delays and wasted time. A unified system keeps all information within the company.

Introducing project management and essential principle templates
Directly on the heels of our new User Interface (UI) released at the end of September and debuting at the RAPS Regulatory Convergence in October, we are proud to announce another HUGE release.
Project management (for large and small teams)
Properly managing registrations across the world with dozens of stakeholders trying to collaborate on critical information is challenging enough, so we just made it easier for enterprise and large teams to manage everything.
The project management features were frequently requested and will set Rimsys up for further development of Key Performance Indicators (KPIs) to monitor the efficiency of your team and the registration process.
It will also allow Rimsys to further expand reporting capabilities and dashboard metrics so you can easily track and analyze data specific to your team, registrations, products, and countries.
- Registration owner - Assign an owner so you know who is responsible for each registration
- Anticipated approval date - Identify an anticipated approval so you can forecast product releases with other departments
- Registration start date - Automatically creates the registration start date so you can monitor exactly how long a registration takes from start to finish.
- Registration lifecycle stages - Whether you are in the discovery, planning, execution or submission stage of the registration process, you can now keep track with your own configurable buckets.
- Kanban boards - Visually see your registrations in each lifecycle stage and transition them into new stages by a simple drag n’ drop interface.
Essential principles (expansion of templates)
We always had a grander plan in mind. Rimsys was originally set up to include the new EU Medical Device Regulation (MDR) 2017/745 Annex I General Safety and Performance Requirements (GSPR) but now supports Essential Principles Templates that include:
- IVDR 2017/746EU IVDR GSPR
- Australian (TGA) Essential Principles
- Japan (PMDA) Essential Principles
- GHTF/SG1/N68:2012 IMDRF Essential Principles
- Directive 2006/42/EC – Machinery Directive
- and more to come!
The essential principles expansion complies with country entrance requirements and will set Rimsys up for further development of correlation tables. What are correlation tables you ask? Think of this….you create the general safety and performance requirements table for the EU MDR, then with a click of a button, you create the essential principle tables that meet the requirements for all other countries. More to come…
What’s next?
Next month, we will be making a few more major announcements that will bring you new and even better features that will drastically create more value to your company and team. We can’t wait to share the news with you…stay tuned!
