


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The beginner's guide to the FDA De Novo classification process
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
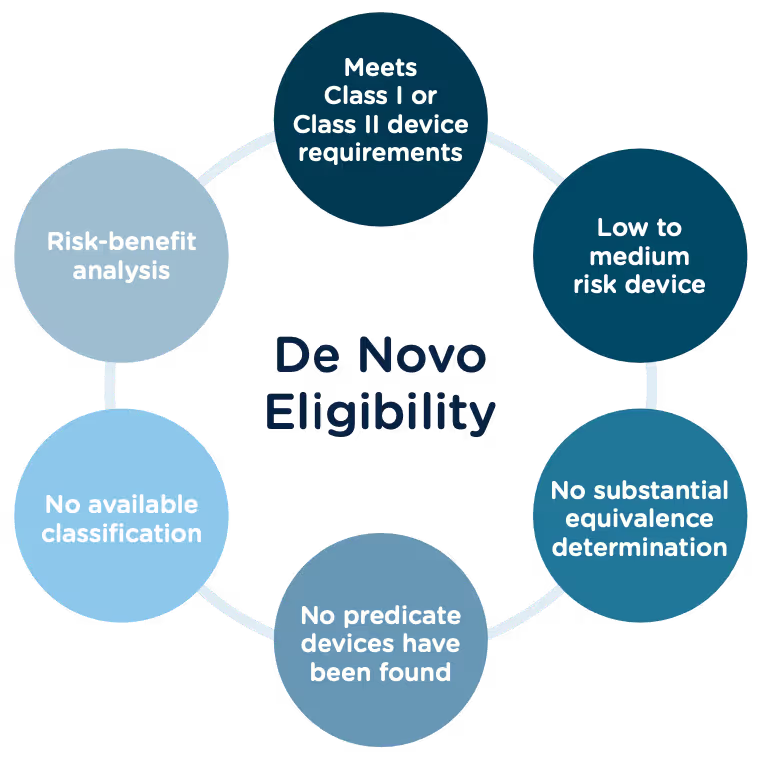
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
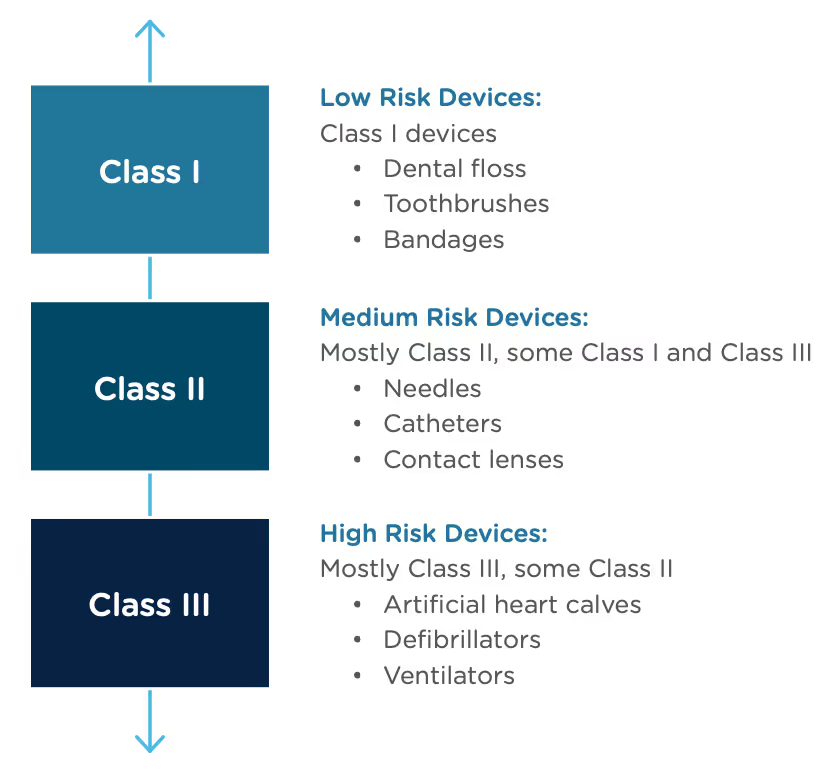
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.




A look at the FDA Total Product Life Cycle Advisory Program (TAP)
The Total Product Life Cycle Advisory Program (TAP) is a voluntary pilot program launched by the FDA’s Center for Devices and Radiological Health (CDRH) in October 2023. The TAP Pilot is one of the commitments between the FDA and industry as part of the MDUFA V reauthorization, which aims to provide faster patient access to safe and effective medical devices, increase innovation, improve patient safety through enhanced surveillance and data collection, and provide a more efficient regulatory process for FDA and industry.
Taking a medical device from concept to commercialization in the United States is often a long and challenging process that involves participation and adoption from FDA, clinicians, payers, and patients. The TAP program is addressing the obstacles that device manufacturers often encounter throughout this process with:
- Early and frequent interactions: FDA will provide more opportunities for sponsors to interact with the agency early in the development process, which can help to identify and address potential issues early on.
- Strategic input from stakeholders: The program will involve input from a variety of stakeholders, including clinicians, patient advocates, and payers, which can help to ensure that the development of new devices is meeting the needs of patients and the healthcare system.
- Proactive, strategic advice from CDRH: FDA will provide proactive and strategic advice to sponsors throughout the development process, which can help to reduce the risk of regulatory delays.
Currently, TAP program membership includes the companies or individuals developing the medical devices, the medical device sponsors, dedicated staff within the CDRH, stakeholders consisting of clinicians, patient advocates, payers, and academic experts, and independent advisors. However, It is unclear if independent advisors will continue to be part of the program as TAP expands. Additionally, specific individuals involved in the TAP program at a given time will vary based on the device being developed and the stage of the development process.
While currently still in its pilot phase, the TAP program is open to a limited number of medical devices. To be considered for the program, device manufacturers must have breakthrough designation with no previous pre-submission meetings. The TAP program started with 15 cardiovascular devices last fall and is now at 31 enrolled devices as of February 2024. Enrollment could reach as high as 60 devices by the end of 2024.
As part of the MDUFA V reauthorization, the TAP Program shares the goal of facilitating the development of high-quality, safe, effective, and innovative medical devices. Additionally, the TAP Program strives to reduce device development time and costs through early and frequent feedback from FDA, increase innovation with more predictable and efficient regulatory pathways for new devices, and improve patient access to new devices.
Overall, the TAP program’s focus is on improving the medical device landscape by addressing various challenges and opportunities throughout the product lifecycle. The program's success will be measured by its ability to expedite development, foster innovation, ensure device quality, and ultimately, improve patient access to these potentially life-changing technologies. For more information about the TAP program and enrollment in it, visit FDA’s website.
_thumbnail.avif)
Are FDA risk classifications and submissions any different for SaMDs? [VIDEO]
The number of software as a medical device (SaMD) is growing and with it are questions about how to effectively obtain market clearance for them. One question we hear often is, “Are FDA risk classifications and submissions any different for SaMDs?” Currently, the FDA is regulating SaMDs the same way it’s regulating traditional medical devices. As a result, you’ll still have the same three risk classifications, Class I, Class II, and Class III.
The submission process is also the same. Most Class I devices are 510(k) exempt, and most class II devices would fall under a 510(k) or De Novo submission depending on whether or not substantial equivalence can be made to another US-marketed device. Most Class III devices require PMA submissions.
There have been discussions about FDA pre-certification programs and following IMDRF guidelines for SaMDs, which would alter the submission process and also the risk stratification of SaMDs. However, none of these discussions have matured. The FDA continues to mirror their risk classification and submission guidelines for SaMDs and traditional medical devices.
Watch the full answer to this question from our recent panel discussion with subject matter expert, Prabhu Raghavan of MDQR Solutions, below.
You can also download the full replay here to get answers to other common SaMD questions such as:
- How is the FDA regulating AI/ML in SaMDs?
- What is a Predetermined Change Control Plan (PCCP) for machine learning-enabled medical devices?
- What cybersecurity considerations sh ould be made when taking SaMDs to market?

An introduction to standards for medtech companies
A standard is a published document that is established by consensus and is approved by a recognized body (ISO, IEEE, UL, etc.). It outlines requirements, specifications, guidelines, or characteristics that are used in a repeated way to ensure that materials, products, processes, and services are developed for a specific purpose. Think of it as a formula for an agreed upon way of doing something that establishes the best way of performing a function. It could be developing a product, managing a process, or even supplying materials to a manufacturer.
Why are standards important?
Standards enable technology to work seamlessly across industries and markets and help to build consumer trust that products and services are designed to work together in an efficient way. They form the fundamental building blocks for product development by establishing consistent requirements that can be universally applied, practiced, and understood. For example:
- Quality standards reduce product failures on the assembly line.
- Environmental standards reduce environmental impacts, reduce waste, and provide sustainability.
- Health and safety standards reduce accidents in the workplace.
- Food safety standards prevent food from being contaminated.
To ensure standards stay relevant and are aligned with technology changes, many standards organizations require that their standards be reviewed periodically and updated as necessary.
Is an industry standard the same as a regulation?
No, but there is a very close relationship between the two. Simply put, a standard is a guideline whereas a regulation includes laws. Industry standards are used voluntarily (although strongly encouraged) while regulations are not voluntary because they are a requirement from a government agency or similar authority, i.e. ISO 13485 is a standard and building codes are regulations.
- Standards are technical documents, driven by consensus that are crafted by experts.
- Regulations are sometimes based on standards, created by a variety of individuals and entities, while overseen by federal, state, and/or municipal authorities.
It's important to note that while standards aren't mandated by law, many regulatory authorities recognize standards and recommend adherence to them in order to promote safety and quality.
What kind of standards are medtech manufacturers responsible for tracking?
The medical device industry has the responsibility for the design and manufacturing of a wide range of products used to diagnose and treat illnesses to improve health in patients. Medical device standards help ensure that a manufacturing or design process can consistently produce the quality required to serve patients and healthcare professionals around the world.
Some of the more common standards used by the medtech industry include, but are certainly not limited to:
ISO 9001 – A general standard (not industry specific) for quality management and implementing a rigorous quality system. For medtech specifically, it helps with the management of the quality control process by helping to keep costs low, improve accountability and simplify regulatory compliance.
ISO 13485 – This standard is designed for medtech specifically and expands on the framework set up by ISO 9001. Compliance with this standard helps with quality control, process validation, and risk management, often referred to as the risk management standard.
ISO 14971 – This standard specifies terminology, principles, and a process for the risk management of medical devices, including software and in vitro. This helps to identify hazards that may be associated with devices and to minimize those risks.
IEC 60601-1 – Medical Electrical Equipment, general requirements for basic safety and essential performance of equipment.
ISO 10993 – Biological evaluation of medical devices that includes biocompatibility testing of materials used to design product parts that would come into contact with a patient, testing for skin sensitization, and irritabilities.
ISO 15223 – Symbols to be used with information to be supplied by the manufacturer. This standard identifies symbols that are globally accepted to be used in a broad spectrum of medical devices. These symbols can be placed on the device, on the packaging, or on any accompanying information such as instructions for use.
ISO 45001 – This standard outlines the requirements for occupational health and safety management systems that can be employed in the medical device industry to help reduce occupational risk.
Where do I get these standards?
Standards used to design and build medical devices need to be purchased, and you must always maintain the most current revision of that standard to ensure proper adherence to it. They can be purchased as electronic copies, or you can request a paper copy for your files. You can purchase them directly from the standard organization (ISO, IEC, ASTM, UL, etc.). There are also organizations services that will provide standards from many organizations, serving as your to be your one-stop shop.
How do I know when standards change?
The best way to manage how you receive information about changes to industry standards would be to implement an electronic standard tracking system. These systems help to:
- Give you early notifications of changes
- Mitigate your company's risk by ensuring you're up-to-date
- Save you time by eliminating the tracking on your own
- Ensure your standards are up-to-date
Using manual processes such as spreadsheets to manage standards updates can be difficult, time-consuming, and lead to compliance risks - especially when a high number of standards and markets are involved. There are a variety of standards management tools to help medtech companies monitor and manage global standards, including Rimsys.
How can Rimsys help?
Rimsys’ regulatory management software offers standards management to help you stay ahead of the mayhem by providing:
- Access to a library of over 1.6 million global standards through a partnership with IHS Markit
- The ability to link standards to individual products to more easily assess the impact of changes across your product portfolio
- Automatic alerts when standards are changed, superseded, or withdrawn to reduce compliance risks and enable faster reaction times
- Bulk updates to your essential principles/GSPR tables when standards change for easier maintenance and compliance
For more information, visit www.rimsys.io/solutions/standards-management.

Why should you invest in your regulatory team? Easy Medical Device podcast interview
Recently, our Founder and CEO, James Gianoutsos, was a guest on an episode of the Easy Medical Device podcast. Hosted by Monir El Azzouzi, a quality and regulatory professional with over 16 years of industry experience, the Easy Medical Device podcast explores a wide range of topics, news, and challenges to help medtech quality and regulatory professionals gain valuable insights that will help them excel in their roles.
In the episode, Why should you invest in your regulatory team?, James and Monir explore the limitations of traditional cost-center approaches to resourcing and preparing budgets for regulatory affairs teams and discuss the benefits of treating regulatory affairs as a revenue function. Hear their thoughts about:
- How regulatory affiars teams are typically structured
- The importance of the RA job function on revenue
- The impact AI will have on regulatory affairs
- How digital tools can enable RA teams
James also provided tips RA professionals can use to convince their stakeholders to invest in regulatory affairs teams. He emphasized that getting buy-in often involves a mindset shift that will change the dynamic of the conversation. For example, when planning for a renewal, think about the financial impacts of missed renewals rather than the sheer volume of renewals you're doing.
When you're trying to convince your leadership team, don't talk about how many renewals I did for this product in a particular month. talk about the dollar figures you saved the company or retained on the market.
For more tips, listen to the full interview on the Easy Medical Device website.

Taking SaMDs to market in the US: How is the FDA regulating adaptive machine learning algorithms?
Rimsys recently held a panel discussion, Taking SaMDs to market in the US. During it, Prabhu Raghavan, Principal at MDQR Solutions, and Rimsys Chief Solutions Officer, Brad Ryba, shared an overview of SaMDs and provided their insights about getting and maintaining market clearance for them in the United States. Topics ranged from FDA risk classifications and submissions, cybersecurity best practices, and machine learning algorithms, which brought about an important question: How is the FDA currently regulating adaptive machine learning algorithms in SaMDS?
Adaptive machine learning algorithms use post-market data in real time and evolve their models based on the data they're consuming. As such, every patient utilizing a device with adaptive machine learning algorithms may have a new model compared to the previous patient. While the FDA doesn't have any formal guidance on the subject just yet, manufacturers can work with the FDA to get a plan in place for maintaining a state of validation post market.
Watch the snippet from the webinar to learn about taking a staged approach with the FDA to get a proper validation plan in place.
To watch all discussion topics, download the webinar replay here.

The five guiding principles for machine learning-enabled medical devices using PCCPs
On October 24th, 2023, the FDA, Health Canada, and the MHRA published a joint document providing harmonization for machine learning-enabled medical devices (MLMD) that use predetermined change control plans (PCCPs). PCCPs are plans proposed by the manufacturer that state the specific modifications to a MLMD, the process for implementing these modifications, and the assessment of impacts from them.
The document details five guiding principles for MLMDs in an effort to set a foundation for PCCPs and encourage collaboration on them. According to the UK government’s website, these principles are:
- Focused and Bounded: Describing specific changes that a manufacturer intends to implement.
- Risk-based: The intent, design, and implementation of a PCCP are driven by a risk-based approach that adheres to the principles of risk management.
- Evidence-based: Demonstrating that benefits outweigh the risks throughout the product lifecycle.
- Transparent: Provide clear and appropriate information and detailed plans for ongoing transparency to all stakeholders, from patients to healthcare professionals.
- Total Product Lifecycle Perspective: Improve the quality and integrity of a PCCP by continually considering the perspectives of all stakeholders.
Here are some examples of how these principles could be applied:
- Focused and bounded: A manufacturer of an MLMD that diagnoses cancer might develop a PCCP to implement a change to the algorithm that improves its accuracy in detecting a specific type of cancer.
- Risk-based: A manufacturer of an MLMD that monitors a patient's vital signs might develop a PCCP to implement a change to the algorithm that reduces the likelihood of false alarms.
- Evidence-based: A manufacturer of an MLMD that delivers medication to patients might develop a PCCP to implement a change to the algorithm that improves the accuracy of the dosage.
- Transparent: A manufacturer of an MLMD might publish a white paper that describes the device's algorithm and how it was developed and tested. The manufacturer might also make available a user manual that provides clear instructions on how to use the device safely and effectively.
- Total product lifecycle perspective: A manufacturer of an MLMD might collect feedback from patients and healthcare professionals on how the device is performing after it is marketed. The manufacturer might also use this feedback to identify and address any potential problems with the device.
The five guiding principles for MLMDs using PCCPs are based on the 10 guiding principles for Good Machine Learning Practices (GMLP) published in 2021, which were designed to help medical device manufacturers develop and deploy machine learning models that are safe, effective, and high quality. Similarly, the goal of these five guiding principles is to help MLMD manufacturers develop and maintain safe and effective products that meet the needs of patients and healthcare professionals. They are also intended to streamline the regulatory process for MLMDs, making it easier for manufacturers to bring new products to market and make updates to existing products in a timely manner.
If you’re looking for additional information about MLMD requirements in the US, join Rimsys and MDQR Solutions for Taking SaMDs to market in the US on Thursday, November 30th, at 1 PM ET. We’ll discuss the various types of SaMDs, considerations to make when obtaining market clearance, and how the FDA is regulating AI/ML in devices. Those interested in attending can register here: Taking SaMDs to market in the US.
