
GUDID is an important source of information as well as a key regulatory requirement for medtech manufacturers who market medical devices, in vitro diagnostics, or medical software in the United States. This article provides an overview of the system, and links to relevant FDA resources you can visit to learn more.
What is GUDID?
GUDID is an acronym for the Global Unique Device Identification Database, a central repository of detailed medical device information created by the FDA. It is often pronounced “Good ID”. The GUDID was implemented as a component of the FDA’s Unique Device Identifier (UDI) requirements, and serves as a digital hub of all the UDI information for all the medical devices that are marketed in the United States.
The GUDID database is designed to help identify and trace all medical devices sold in the U.S., and provides detailed specifications about each device including manufacturer and production information, intended use, safety, and storage and handling requirements. The database is accessible to regulators, manufacturers, healthcare providers, insurers, and the public at large.
GUDID history
The GUDID was implemented as a part of the FDA’s UDI system. This system requires that each medical device have a unique identification code that is included in the device label (printed on the device itself or its packaging) in both machine and human readable format. An example of a UDI code is included below. The UDI code contains information about the device, the manufacturer, and when/where the device was manufactured.
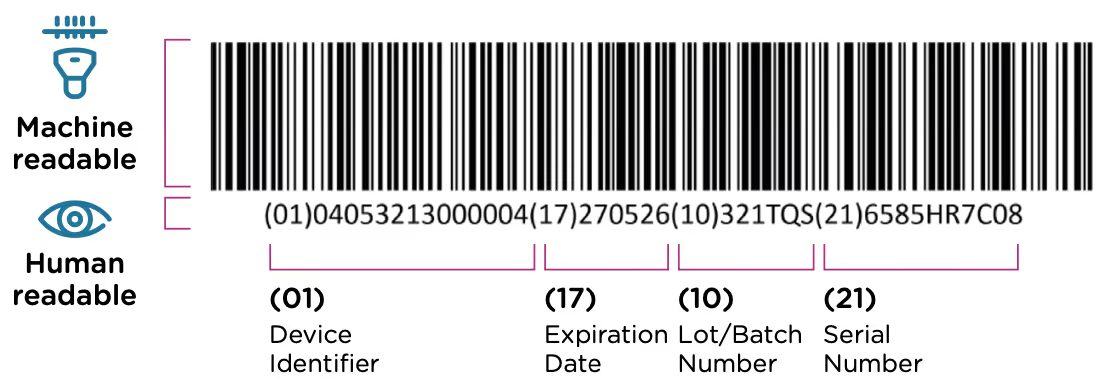
The FDA’s UDI program was established in 2013, when a rule was issued requiring all medical devices to carry a UDI by 2020. The GUDID database was included with the same regulation, and manufacturers were required to submit all of their UDI information electronically to this database as the requirements came online for different device classes. The overall UDI requirements rollout had the following timeline:
The GUDID database was launched ahead of the first device deadline in December, 2013, and the public access portal AccessGUDID went live in May, 2015.
Who should submit data to the GUDID?
The FDA specifies that the GUDID submission is the ultimate responsibility of the “device labeler”. This is the entity/company who is identified on the device’s label (which also contains the UDI code). So the same entity that attaches the UDI to the device, is also responsible for the electronic GUDID submission. In almost all cases this is the manufacturer of the device, however, it can be the U.S. distributor for the product if they are named on the product’s label.
What data must be submitted to the GUDID?
The information submitted to the GUDID includes all of the device information that is in the UDI code along with additional information about product distribution, product and packaging size, sterilization, and storage and handling instructions. The following information is required to be included with each submission:
- Device identifier information - This includes the device identifier (the first part of the UDI code), a detailed device description, and information about the labeler including the DUNS code, and company name and address.
- Commercial distribution - This includes the distribution status—whether or not the device is in commercial distribution, and the distribution end date—when the device will no longer be distributed.
- Alternative identifiers - If the device has another DI, either a direct marketing DI, a distinct packaging DI, one from another issuing agency, or one that was used previously, this information must be provided.
- Customer contact information - A phone and email address for patients or consumers who have questions about the device.
- FDA codes and listing number - If the device completed a pre-market authorization (PMA) that should be included as well.
- Manufacturing information - Manufacturing date, lot or batch number, serial number, and expiration date for the device.
- Latex information - Whether or not the device or its packaging contains rubber components.
- Device dimensions - What is the clinically relevant size and unit of measure for the device.
- Storage and handling - Requirements and parameters for storage including temperature, humidity, and pressure.
- Sterilization - Whether the device is packaged as sterile or requires sterilization prior to use.
The FDA provides a detailed spreadsheet of data requirements that you can use to prepare your submission.
Creating a GUDID submission
In addition to gathering the required information (and obtaining a UDI code for your device) there are several additional steps to complete in order to create a GUDID submission for your product. First, if you don’t have one, you’ll need to create a GUDID account. The FDA allows you to request an account online. Note that you will need to have a DUNS number for your business. If you don’t have one you can request one from Dun & Bradstreet at no cost.
There are two ways that you can enter your submission. You can do this online through the GUDID Web application. The FDA also allows you to submit your GUDID information all at once using an XML file that complies with Health Level 7 (HL7) Structured Product Labeling (SPL) formats. These submissions are made via the FDA Electronic Submissions Gateway, and require you to set up a gateway account.
In addition, some software providers (like Rimsys) include the ability to make electronic GUDID submission directly from their tools. They provide a system to organize and manage UDI data for the US and other countries, and can ensure that GUDID information for your products is kept up to date.
The global proliferation of UDI regulations
The GUDID was one of the first public databases of medical device information, but many additional countries and regions have followed suit. The European Union, China, South Korea, and Taiwan have all introduced UDI databases and requirements that manufacturers submit records for all of their products sold in-market, and ensure that they are kept up to date.
For more information about global UDI programs and timelines, see our UDI quick reference guide. And you can find more detailed information about the specific requirements in the EU and China in our Ultimate guide to the MDR/IVDR UDI and Ultimate Guide to the China NMPA UDI requirements ebooks.


GET IN TOUCH






.avif)
