


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR)
This article is an excerpt from The ultimate guide to the EU MDR and IVDR general safety and performance requirements (GSPR) ebook.
Table of contents
- Overview
- Terminology
- EU MDR/IVDR Annex I
- EU MDR/IVDR Annex II
- Proactive Monitoring & Maintenance
- Comparison Table: EU MDR/IVDR Annex I GSPRs vs EU MDD/IVDD Annex I Essential Principles
With the initial rollout of the European Medical Device Regulation (MDR) complete, medical device companies are shifting focus to the sister In Vitro Diagnostic Regulation (IVDR) which has rolling effective dates starting in May 2022. Like the MDR, the IVDR also includes new General Safety and Performance Requirements (GSPR). The expanded 2nd edition of this ebook includes a detailed summary of the IVDR GSPR regulations in addition to those of the MDR. It provides you with practical guidance on how to meet the GSPR requirements for all types of medical technology products. This ebook, however, should not take the place of reviewing the actual regulations and consulting regulatory experts when needed
Timeline
The EU MDR submission became mandatory from the previous MDD directive on May 26, 2021, and the EU IVDR effective date is quickly approaching. In fact, all submissions for new devices under the new EU IVDR must be implemented no later than May 25, 2022. Below is a high-level overview of key dates for both regulations.
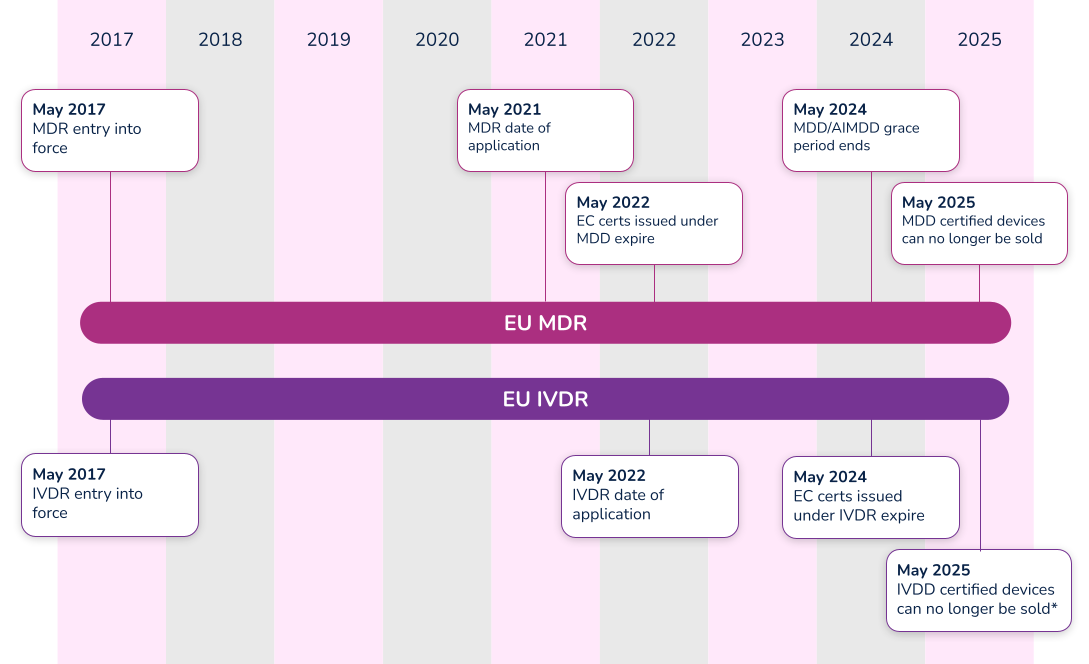
*Note that the timeline for compliance was extended in 2021. Class D (high-risk) devices have until 2025 to comply with IVDR, while Class C devices have until 2026. Class B and Class A sterile devices have until 2027 to comply with IVDR.
What’s the difference between Essential Requirements, General Safety and Performance Requirements (GSPR), and Essential Principles. In order to have a meaningful dialogue, let’s first discuss the three (3) main terms used in the industry.
#1 Essential requirements
The ‘Essential Requirements’ is the backbone for establishing conformity with the Medical Device Directive (MDD 93/42/EEC) and the Active Implantable Medical Device Directive (AIMDD 90/385/EEC). Detailed within Annex I of the MDD and AIMDD, the ‘Essential Requirements’ laid out the requirements that devices must meet in order to state compliance to the directives. With the implementation of the new EU Medical Device Regulation (MDR 2017/745), the ‘Essential Requirements’ will become superseded by the new EU MDR General Safety and Performance Requirements (GSPRs).
#2 Essential principles
The IMDRF laid out Essential Principles requirements in a document entitled Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices. From a high-level perspective, three basic tenets make up these ‘Essential Principles’:
- A device must be designed to be safe and perform effectively throughout its lifecycle.
- Device manufacturers must maintain all design characteristics.
- Devices must be used in a way that is consistent with how it was designed.
Many countries use the term ‘Essential Principles’ when compiling the documentation required to determine compliance to the law. For instance, the Australian Therapeutic Goods Administration (TGA) uses the term ‘Essential Principles Checklist’. Regardless of the term used, Essential Principles are of similar nature and overlap many of the Essential Requirements and new GSPRs.
#3 General safety and performance requirements (GSPR)
As of May 26, 2021, medical device manufacturers must start to comply with Annex I – General Safety and Performance Requirements (GSPRs) of the new EU Medical Device Regulation (MDR 2017/745). GSPRs are specific to the European MDR and IVDR. If you hear any other term (i.e. Essential Principles), it most likely means it is not referencing the European market.
Annex I of the EU MDR and IVDR details the specific requirements of the General Safety and Performance Requirements (GSPRs). The GSPRs are broken down into three (3) chapters in Annex I, MDR 2017/745 and IVDR 2017/746:
- Chapter 1 - General requirements
- Chapter 2 - Requirements regarding design and manufacture
- Chapter 3 - Requirements regarding the information supplied with the device
Chapter 1 - General requirements
Both the EU MDR and the EU IVDR outline General Safety and Performance Requirements (GSPRs) in great detail for medical device designers and manufacturers. The general requirements for each are almost identical and consist of the following:
- Devices must perform in a way that aligns with the intended design.
- They must not compromise the health or safety of a patient, user, or any other person associated with the device.
- Risks must be reduced as much as possible, but not so much that they negatively affect the risk-benefit ratio.
- Device manufacturers must implement and maintain a thorough, well-documented, and evaluative risk management system that continues to be updated throughout the life cycle of a device.
- Manufacturers and designers must include any necessary measures for protecting users in cases where risks cannot be completely eliminated.
- Manufacturers must provide users with information about any potential risks that remain. This information must be clear, easy to understand, and considerate of the users’ technical knowledge level, use environment, and any applicable medical conditions.
- Devices must withstand the stresses of normal use for the duration of their lifecycle. Devices must be designed, manufactured, and packaged in a way that protects them from damage during transport and storage.
- When it comes to risks and negative side effects that are known and foreseeable, designers and manufacturers must make every effort to minimize negative outcomes. They must also ensure that potential risks are acceptable when compared to the potential benefits of a device to its users.
Chapter 2 - Requirements regarding design and manufacture
The GSPRs also provide key details regarding specific information about the performance, design and manufacture of medical devices. As it relates to design inputs, the MDR and IVDR GSPRs provide highly detailed requirements relating to a device’s technical information. Further detail can be found in the comparison tables in Appendix A and Appendix B, where we have compared MDR to MDD and IVDR to IVDD.
Chapter 3 - Requirements regarding the information supplied with the device
The final key area of governance within the GSPRs relates to specific information a manufacturer must supply with a device. The general requirements for this information states that, “Each device shall be accompanied by the information needed to identify the device and its manufacturer, and by any safety and performance information relevant to the user, or any other person, as appropriate.” The requirements provide further detail as far as location - specific information that must be provided on the following:
- The device label includes its UDI.
- The user instructions.
- The packaging of a device that is intended to maintain its sterile condition.
Medical devices are subject to significant regulations and a full understanding of EU MDR and/or IVDR labeling as defined in Annex 1 Chapter 3.
In addition to the specific requirements identified within Annex I of the EU MDR and IVDR, Annex II, Technical Documentation, identifies additional requirements. Specifically, in both EU MDR and IVDR’s Section 4 – General Safety and Performance Requirements it states:
“the documentation shall contain information for the demonstration of conformity with the general safety and performance requirements set out in Annex I that are applicable to the device taking into account its intended purpose, and shall include a justification, validation and verification of the solutions adopted to meet those requirements. The demonstration of conformity shall include:
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
(c) the harmonised standards, CS or other solutions applied; and
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonised standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation.”
Let’s break this down into each part.
Requirement
(a) the general safety and performance requirements that apply to the device and an explanation as to why others do not apply;
What needs to be documented for the requirements that apply or the requirements that do not apply?
Each and every section of the EU MDR GSPR or EU IVDR should be assessed in its own right as it pertains to your medical device. When a requirement applies, a simple statement may be made that this requirement applies to the device. In practice this is often achieved using a checklist or table, with a column for applicability and a Yes/No answer against each requirement. When a requirement applies, you can move on to the other parts of demonstrating conformity regarding methods used and standards applied.
When a requirement is not applicable, a statement must be made to that effect, i.e. a ‘No’ in the applicability column. Additionally, it must be fully and properly justified. Such a justification may be something like ‘The device is not powered and is therefore not an active device. This requirement does not apply.' The justification should clearly state why the requirement has been deemed not to apply so that your notified body can understand your reasoning
Requirement
(b) the method or methods used to demonstrate conformity with each applicable general safety and performance requirement;
What is meant by “method or methods used”?
This relates to the way you complied with that GSPR requirement, historically it would be listed as a standard or other documentation reference that you have applied to demonstrate compliance, however, the question of ‘method or methods used’ is new to the MDR and it is expected that a verbal description be provided such as:
i. Risk analysis weighed against clinical evaluation benefit
ii. Performance intended demonstrated by design requirements, verification and validation
Requirement
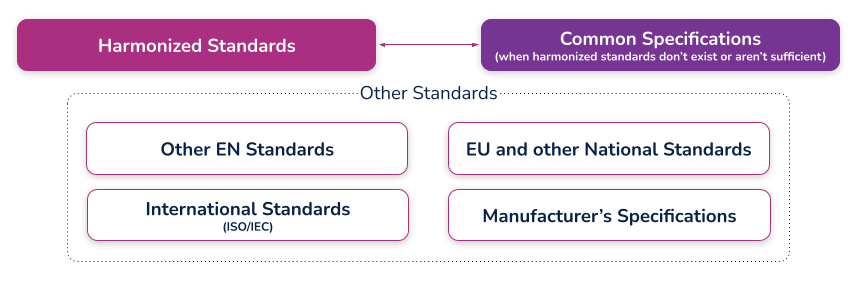
(c) the harmonized standards, common standards (CS) or other solutions applied;
What are harmonized standards, common specifications (CS), and “other solutions”?
Harmonized standards
These are standards that have been specifically developed and assessed for compliance to a regulation or directive. They are published in the Official Journal of the European Union (sometimes just referred to as ‘the OJ’) and if you comply with these standards then there is a ‘presumption of conformity’ with that directive or regulation to which they have been harmonized. These harmonized standards can only be created by a recognized European Standard Organization (such as CEN or CENELEC). When a standard is harmonized, an annex is added that describes how the standard conforms to the directive or regulation. When using harmonized standards, you should make sure that you understand how the standard conforms so that you do not claim compliance when the standard either does not meet that requirement or only partially meets that requirement.
If a standard does not meet a certain requirement of the directive or regulation, or indeed only partially meets it, then you must employ additional mechanisms for compliance. If a harmonized standard meets part of a directive or regulation, then by complying with that standard you also fully meet the corresponding requirement(s) The list of harmonized standards continues to grow - refer to the “Healthcare Engineering” section of the European Commission’s Harmonized Standards page for current information. In this case, using an MDD harmonized standard and documenting a justification for doing so (i.e. how you believe the standard demonstrates compliance with the GSPRs), should provide sufficient evidence
Common specifications
Common Specifications (CS) are a new concept in the MDR. They allow the European Union to add additional requirements that must be met in order to claim compliance where harmonized standards do not exist or where relevant standards are considered insufficient. The definition of a Common Specification is:
‘A set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device, process or system.’
Requirement
(d) the precise identity of the controlled documents offering evidence of conformity with each harmonized standard, CS or other method applied to demonstrate conformity with the general safety and performance requirements. The information referred to under this point shall incorporate a cross- reference to the location of such evidence within the full technical documentation and, if applicable, the summary technical documentation;
What is the expectation for incorporating a "cross-reference to the location of such evidence within the full technical documentation"?
This means that someone looking at the document should be able to identify exactly where in the technical documentation that the compliance evidence can be found. For example, this may refer to test reports and their exact location, or it could even reference locations within a large document, depending on the GSPR and your particular documentation. (i.e. if you have included usability risks as part of a larger risk assessment, you may need to say ‘See Technical File XXX, Section XX, Doc RMF001 rev 3 lines 65-78’). In other cases it could just mean the whole document reference, i.e. Have you done risk management? – then yes, it is RMF001 rev 3. What the specific reference actually is depends on how you have managed your technical documentation and how defined it is (i.e. separate reports or one big one). There should be no ambiguity as to where the document is located
An example of a completed GSPR checklist could look something like this (applicable and nonapplicable examples are shown):
Specification developers and manufacturers must continually maintain their technical documentation to stay compliant. Part of this process is to ensure that they take into account the "generally acknowledged state of the art".
Proactive monitoring
'State of the art'
There is no formal definition of ‘state of the art’ within the EU MDR or IVDR, although it is mentioned many times. ‘State of the art’ is an ongoing debate; however, it generally means that it embodies what is currently and generally accepted as good practice in the medtech industry. The ‘state of the art’ does not necessarily imply the most technologically advanced solution.
One consensus on state of the art is being up to date and compliant with the current and in effect standards that are applicable to your device. This means that if a standard is updated that your medical device is compliant with, you must evaluate that update to ensure that it would meet the EU MDR or EU IVDR ‘state of the art’ requirement. This is not a new requirement from the EU MDD but it is spelled out more clearly in the EU MDR.
The specification developer or manufacturer is ultimately responsible for determining if the updated standard applies or does not apply to their device(s). Either way, the justification should be documented within a gap analysis.
Monitoring for changes
Of course, 'state of the art' only applies if you actually know if something changed. This is why you need to develop a process for monitoring the standards that compliance is claimed. Every single standard that is associated with your technical documentation must be actively monitored, reviewed, and reported on.
If you have a product on the market and need a better way to monitor and maintain your General Safety and Performance Requirements (GSPR) or Essential Principles, Rimsys can help. Rimsys digitizes and automates GSPR and Essential Requirements so you can dynamically update and proactively monitor changing standards and evidence files.
When a standard or evidence file changes, you will automatically be notified and can update one GSPR or all of your GSPRs as applicable with a single click of a button. If additional information is needed, such as testing, it’s also invaluable to ensure that all devices are identified. What used to take weeks of manual, error-prone administrative tasks is now done in seconds within a fully validated, secure, maintenance-free, cloud-based solution
Maintenance
Maintaining and updating your technical documentation is generally the hardest part of staying compliant. Robust processes must be established to ensure nothing slips through the cracks and show up as nonconformances during regulatory audits.
Gap analysis
In addition to meeting the ‘state of the art’ requirements and the continuous proactive monitoring of standards, once a change has been detected that affects the technical documentation, a proper and thorough gap analysis must be completed.
The gap analysis between the old versions and the new versions, or an evaluation of a brand new standard, must occur and be properly documented. The gap analysis should detail what is applicable and what is not applicable, with your supporting justification.
If something within the new or revised standard was applicable to your device, additional engineering testing, documentation, justification, and, in some instances design changes, may be needed to ensure compliance
GSPR updates
Once the gap analysis has been properly documented, specification developers and manufacturers must update their GSPRs.
These updates include finding the withdrawn or superseded standard or evidence file throughout each row within your GSPR table, for every single device on the market on which this change is applicable. This could be one table or dozens of tables depending on the complexity of the products and your product mix.
Without a holistic RIM system to help you, this is an error-prone process as is it tedious, administrative, and extremely easy to miss an inappropriate referenced standard or evidence file.
Extreme diligence on the regulatory or engineering team must occur to ensure these critical updates to the GSPRs are not missed and a gap analysis must be properly referenced throughout. Any justification for including or excluding a new standard or evidence file will be scrutinized by regulatory auditors, and without proper maintenance, may lead to additional review time.
To continue reading this eBook including Comparison Table of the EU MDR Annex I GSPR vs. the EU MDD Annex I Essential Requirements, please register to download the full version.

The beginner's guide to the FDA PMA submission process
This article is an excerpt from The beginner's guide to the FDA PMA submission process ebook.
Table of Contents
- Introduction
- PMA basics
- FDA interactions
- Contents of a traditional PMA submission
- PMA supplements and amendments
- PMA Quality Management System (QMS)
- Review process and timeline
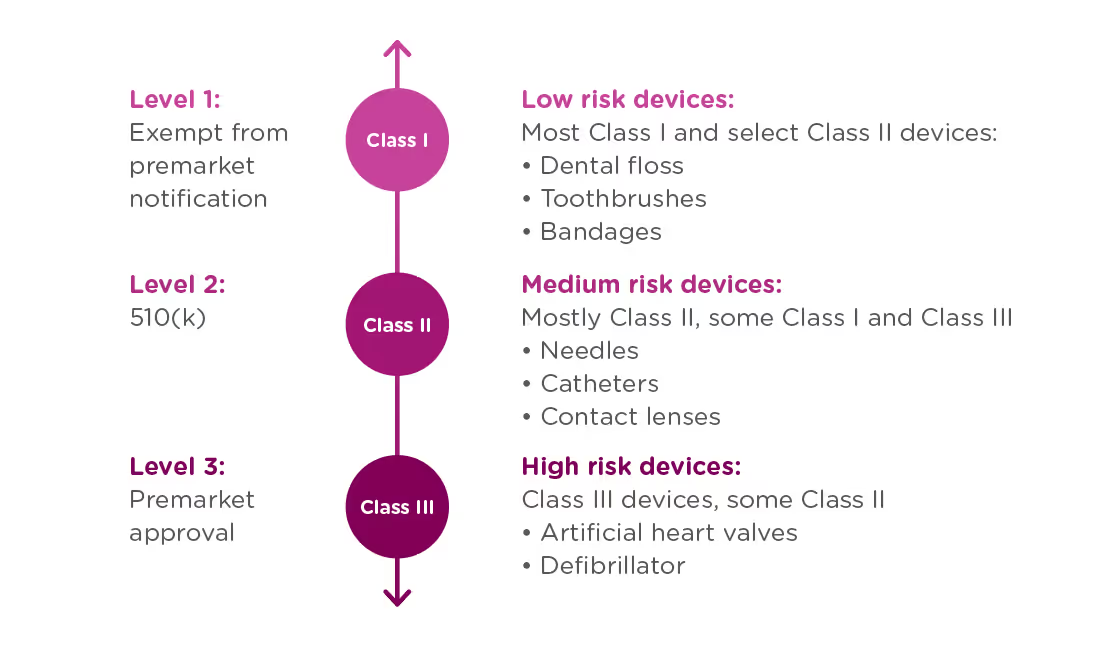
If your organization is planning to market a new medical device in the United States, you first need to determine which regulatory class the device falls under. The vast majority of medical devices regulated by the FDA are either Class I or Class II medical devices, requiring a 510(k) premarket notification or a simple registration if exempt from 510(k) requirements. However, if your device sustains or supports life, is implanted, or presents a “potential unreasonable risk of illness or injury,” your device is likely a Class III device which will require Premarket Approval (PMA) from the FDA before it can be marketed in the United States. Novel devices, for which there are no existing substantially equivalent devices, are automatically classified as Class III as well. Novel devices with a lower risk profile, however, may qualify for the De Novo process instead of the PMA. Just 10% of devices regulated by the FDA are Class III devices.
This ebook provides an overview of the PMA process and its requirements, but it is not designed to be the only resource used in compiling a PMA submission. The FDA provides significant documentation on this process, starting with the regulation governing premarket approval that is located in Title 21 Code of Federal Regulations (CFR) Part 814.
FDA: Background and device oversight
Before we explain what a PMA is, let’s first talk generally about the Food and Drug Administration (FDA) and device oversight. The FDA is the U.S. governmental agency responsible for overseeing medical devices, drugs, food, and tobacco products. When it comes to medical devices, the FDA’s mission is to “protect the public health by ensuring the safety, efficacy, and security of...medical devices.” At the same time, the FDA also has an interest in “advancing public health by helping to speed innovations.” In other words, the FDA’s goal is to make sure devices are safe and effective for public use, while also ensuring that devices have a quick and efficient path to market.
In order to achieve this balance of safety and efficiency, the FDA has three different levels of oversight depending on the risk level of the device: (1) exempt from premarket notification, (2) Premarket Notification, also known as 510(k), and (3) Premarket Approval (PMA).
When is a PMA required?
The PMA process is the most stringent regulatory process for medical device approval under the FDA and applies to almost all Class III devices. To determine whether your device requires a PMA, you must first Classify your device by searching the Product Classification Database. The database will provide you with similar devices; their name, classification, and link to the Code of Federal Regulations (CFR) if applicable.
- If a substantial equivalent is found in the Product Classification Database with a submission type of 510(k), you should submit a 510(k), not a PMA.
- If the product classification database identifies your device as Class III and/or requiring a PMA - you should submit a PMA.
- If your device involves a new concept and does not have a classification regulation in the CFR, the database will list only the device type name and product code. In this case, the three-letter product code can be used to search the PMA database and the 510(k).
- If your device cannot be found in the product classification database because it is a new type of device and should be classified as a Class III device because of the level of risk it presents*.
Class III devices support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential and unreasonable risk of illness or injury.
Note that if your device is a new concept without a substantial equivalent, but does not present the level of risk of a class III device, it may be eligible for the De Novo process as a class I or class II device.
PMA vs 510(k)
Not only are PMA and 510(k) processes applicable to different types of devices, they have different purposes.
510(k): A 510(k) is intended to demonstrate that the device for which approval is being sought is as safe and effective as a currently marketed device that does not require a PMA.
PMA: A PMA is intended to prove that a new device is safe and effective for the end user. A PMA is much more detailed and in-depth than a 510(k). Device manufacturers are typically required to present human clinical trial data, in addition to laboratory testing data.
The difference in complexity between a PMA and 510(k) also affects the time needed to process the submissions. The FDA typically accepts or rejects a 510(k) submission within 30-90 days, at which point the device is posted to the FDA’s 510(k) database. A PMA submission can take up to 180 days to be processed, at which point the FDA can approve or deny the application. The FDA may also issue an “approvable” or “not approvable” letter, which the applicant can choose to respond to, thereby adding time to the submission process.
PMA application methods
There are a number of types of PMA application methods. While most devices which require a PMA will follow the traditional process, be sure to verify that you are using the correct application process to maximize your chances for success and avoid unnecessary delays:
Traditional PMA
The most common method for attaining FDA clearance for Class III devices, the traditional PMA is the appropriate option for most devices that have completed clinical testing.
Modular PMA
The modular PMA is the appropriate application method for devices that have not yet completed clinical testing. Applicants complete individual “modules,” with final confirmation granted once all sections are completed. For additional information on specific requirements of a modular PMA, read the FDA’s Premarket Approval Application Modular Review.
Product Development Protocol
Use the Product Development Protocol (PDP) with medical devices that are based on well-established technology. The PDP process for gaining market approval merges the clinical evaluation and development of information, and involves an agreement between the manufacturer and the FDA. The process provides the advantage of early predictability for the manufacturer and allows early interaction that can identifyFDA concerns as soon as possible in the development process. Because the PDP identifies the agreed upon design and development details, a completed PDP is considered to have an approved PMA. For additional information, read more about the FDA’s PMA Application Methods.
Humanitarian Device Exemption
A Humanitarian Use Device (HUD) is specifically defined as a device intended to benefit patients that are affected by a disease or condition that affects less than 8,000 individuals in the U.S. per year. TheHumanitarian Device Exemption (HDE) approval process is designed to encourage clinical activity around rare conditions, and does have certain restrictions, including:
- After receiving HDE approval, a HUD is eligible to be sold for profit only if the device is intended to address a disease or condition that occurs primarily in pediatric patients, or occurs in pediatric patients in small numbers.
- If an HDE is approved to be sold for profit, the FDA will determine an annual distribution number(ADN). Any devices sold beyond the ADN limit are required to be sold for no profit.
For more information see the FDA’s explanation of the Humanitarian Device Exemption.
CBER Submissions
There are two centers within the FDA responsible for evaluating medical devices. While the majority of devices will go through the Center for Devices and Radiological Health (CDRH), some will be managed by The Center for Biologics Evaluation and Research (CBER). CBER regulates medical devices related to blood and cellular products, including blood collection and processing procedures as well as cellular therapies. This ebook focuses on submissions made through the CDRH, but you can view CBER Regulatory Submissions – Electronic and Paper for more information on the CBER process.
To continue reading this eBook, including a walk through of the different types of required and optional FDA meetings and communications, a detailed list of the contents of a traditional PMA submission, and an overview of quality management system requirements, please register to download the full version.

An overview of 21 CFR Part 11 regulations for medical device companies
What is 21 CFR Part 11?
21 CFR Part 11 refers to the federal regulation that address electronic records and electronic signatures associated with FDA requirements. This single, relatively small, part of the Code of Federal Regulations is extremely significant for companies with FDA-regulated products because it impacts every document signature, electronic file, and FDA submission. Codified in 1997, interpretations of this FDA-issued regulation continue to be debated and re-evaluated as the technology supporting electronic records and signatures changes. In this article, we’ll discuss the regulation and generally accepted interpretations.
Note that discussions and statements in this document are our observations only and should not be taken as fact. You can refer directly to the regulation here.
Part 11: General Provisions
The General Provisions section of 21CFR11 addresses the scope of the regulation, when and how it should be implemented, and defines some of the key terms used. It states that the purpose of Part 11 is to define the criteria under which electronic records, electronic signatures, and handwritten signatures attached to electronic records are equivalent to, and as reliable as, handwritten signatures on paper documents.
Fundamentally, any record that is maintained, used, or submitted under any FDA records regulation is subject to Part 11, and the FDA will accept electronic records in lieu of paper records if an organization can prove that their records and systems meet the Part 11 requirements.
The General Provisions subpart also sets forth a number of definitions, and we’ve listed the ones that are most significant to our discussion here:
- Closed System: A computer system or software whose access is controlled by the same people who are responsible for the information stored in the system. Because the opposite of a closed system, and “open system,” is subject to additional scrutiny be sure that you are able to thoroughly explain and provide documentation for a decision to classify your system as a “closed system.”
- Open System: A computer system or software whose access is not controlled by the same people who are responsible for the information stored in the system.
- Digital Signature: An electronic signature created in a manner that can be verified, ensures the identity of the signer, and maintains the integrity of the document and signature. This often involves the use of cryptography and/or biometric data.
- Electronic Signature: Symbols that represent a legally binding equivalent to an individual’s handwritten signature (as adopted and authorized by the signer).
Part 11: Electronic Records
The Electronic Records section sets forth the requirements for administration of closed and open electronic record-keeping systems, then discusses signature manifestations and requirements for establishing a link between signatures and records.
Part 11 defines a “closed system” as any computer system in which the users controlling access to the system are the same people who are responsible for the data in the system. Today, most systems can be classified as closed systems, but take special care to document control procedures around software that is hosted offsite or classified as a SaaS solution.
This section of the regulation deals with the controls that need to be in place for all applicable electronic record systems by defining:
- Procedures to ensure that all electronic records are authentic, have integrity, and can ensure confidentiality (where that is appropriate).
- Validation requirements for systems that maintain electronic records to ensure that all records are accurate, reliable, and that the system performs consistently according to regulatory requirements.
- Audit trail requirements for all regulated records to ensure a complete history of all changes to records are maintained.
- Controls around system access and document signatures.
Part 11: Electronic Signatures
The Electronic Signatures section defines the components of electronic signatures and the required controls and procedures necessary for using them.
In general, an organization must be able to demonstrate that electronic signatures:
- Are unique to each individual, and that the individual assigned an electronic signature has had their identity and level of authorization verified.
- Must be based either on biometric data (such as fingerprints) or made up of two distinct pieces (ie: a User ID and password)
- Require appropriate controls to ensure that they are verified periodically, cannot be used by someone other than the intended user, and are immediately deactivated if compromised in any way.
Practical application of 21CFR Part 11 for regulatory affairs professionals
21 CFR Part 11 is a critical regulation, and one that can be open to interpretation. Below, we cover some of the key areas that should be of concern for RA professionals. This is an overview of key areas only, and should not be taken as complete instruction or guidance for 21CFR part 11 compliance.
System compliance and validation
Any system that you are using to store electronic records that fall under FDA regulations needs to be compliant with Part 11. This includes everything from spreadsheets to full-featured RIM and document management systems.
Software vendors will often document how their systems are developed to be compliant, and may even support system validation during implementation - but it is ultimately the responsibility of the user organization to ensure that their systems and processes are compliant with Part 11. System validation is the process of documenting that your system meets all of the Part 11 requirements. Software vendors can support this process by ensuring that their systems are built on a highly secured infrastructure that can be demonstrated and proven.
The Rimsys system was built from the ground up to meet the stringent requirements of not only 21 CFR Part 11, but other industry standards and good practices guidelines (GxP). We have put in place a rigorous validation program, built by industry experts and supported by a secure and well-documented infrastructure. For more information, visit the Rimsys Security and Privacy page.
Audit trails
Audit trails are the required system logs that track the who, when, and what of every change made to data that falls under Part 11. Audit trails should be generated and time-stamped by the system, with no ability for users to change that information. Audit trails serve two purposes under 21 CFR Part 11:
- To demonstrate that documented policies and procedures are being followed, including that only users with the appropriate authority are managing data.
- To prove that data retention policies are being adhered to (see below).
At any time, you should be able to view the history of any record, from a Design History File to a submission document, in order to determine what changes have been made, when they were made, and by whom.
Record retention
21 CFR Part 11 specifies that electronic records must be protected and readily available throughout the defined record retention period. Additionally, 21 CFR Part 820 specifies that records related to the quality, manufacturer, regulatory submissions, or any other data that falls under FDA regulation, should be maintained for the life of the medical device and for a minimum of two years from the date of first commercial distribution. This is often referred to as “cradle to grave” tracking.
This means that regulatory professionals need to not only be aware of their company’s record retention policy, but need to ensure that any system being used to track regulatory submissions or other data subject to audit meets Part 11 and Part 820 requirements. Note that record retention requirements apply also to paper records where they are the source document.
Electronic and digital signatures
An important piece of 21 CFR Part 11 is its definition of electronic and digital signatures. “Electronic signature” is used to define any set of symbols that are used in place of a handwritten signature, whereas a “digital signature” is an electronic signature based on methods that ensure the identity of the signer where the integrity of the data can be verified. A digital signature can be based on biometric data (such as fingerprints) or secure user IDs and passwords that are controlled to ensure only one authorized user can use the signature.
As a regulatory affairs professional, you should ensure that:
- Everyone on your team who needs to sign documents has their own unique digital signature and understands the importance of protecting it. Sharing of electronic credentials is a common FDA audit observation. Also ensure that users who are not required to sign documents have appropriate access to data to discourage other users from sharing login credentials with them.
- You are following your company’s policies concerning electronic signature audits so that passwords remain updated and strong and signatures are revoked when a user leaves or changes positions.
- You immediately report any possible loss, theft, or sharing of user credentials or devices that generate identification codes.
While 21 CFR Part 11 is usually considered more of a “quality regulation,” it is important that regulatory teams within medical device organizations fully understand this regulation and its compliance implications. To learn more about the regulations, click below to read our regulatory brief.


GUDID: An overview of the FDA's Global Unique Device Identification Database
GUDID is an important source of information as well as a key regulatory requirement for medtech manufacturers who market medical devices, in vitro diagnostics, or medical software in the United States. This article provides an overview of the system, and links to relevant FDA resources you can visit to learn more.
What is GUDID?
GUDID is an acronym for the Global Unique Device Identification Database, a central repository of detailed medical device information created by the FDA. It is often pronounced “Good ID”. The GUDID was implemented as a component of the FDA’s Unique Device Identifier (UDI) requirements, and serves as a digital hub of all the UDI information for all the medical devices that are marketed in the United States.
The GUDID database is designed to help identify and trace all medical devices sold in the U.S., and provides detailed specifications about each device including manufacturer and production information, intended use, safety, and storage and handling requirements. The database is accessible to regulators, manufacturers, healthcare providers, insurers, and the public at large.
GUDID history
The GUDID was implemented as a part of the FDA’s UDI system. This system requires that each medical device have a unique identification code that is included in the device label (printed on the device itself or its packaging) in both machine and human readable format. An example of a UDI code is included below. The UDI code contains information about the device, the manufacturer, and when/where the device was manufactured.
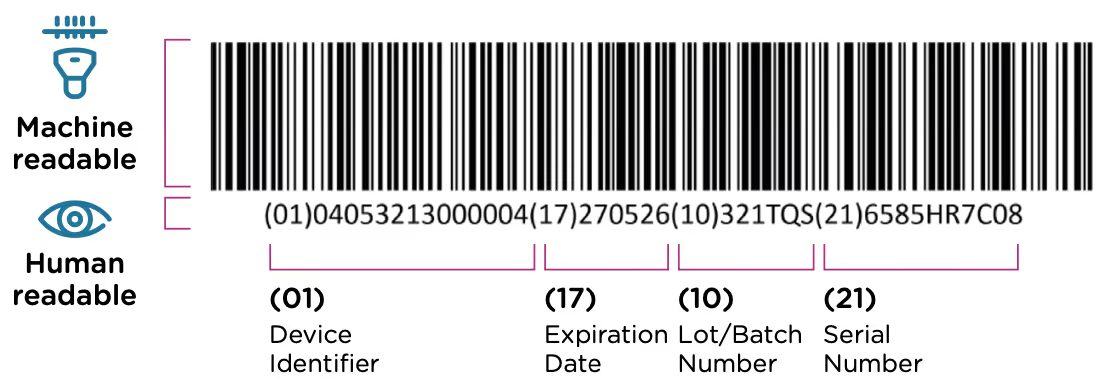
The FDA’s UDI program was established in 2013, when a rule was issued requiring all medical devices to carry a UDI by 2020. The GUDID database was included with the same regulation, and manufacturers were required to submit all of their UDI information electronically to this database as the requirements came online for different device classes. The overall UDI requirements rollout had the following timeline:
The GUDID database was launched ahead of the first device deadline in December, 2013, and the public access portal AccessGUDID went live in May, 2015.
Who should submit data to the GUDID?
The FDA specifies that the GUDID submission is the ultimate responsibility of the “device labeler”. This is the entity/company who is identified on the device’s label (which also contains the UDI code). So the same entity that attaches the UDI to the device, is also responsible for the electronic GUDID submission. In almost all cases this is the manufacturer of the device, however, it can be the U.S. distributor for the product if they are named on the product’s label.
What data must be submitted to the GUDID?
The information submitted to the GUDID includes all of the device information that is in the UDI code along with additional information about product distribution, product and packaging size, sterilization, and storage and handling instructions. The following information is required to be included with each submission:
- Device identifier information - This includes the device identifier (the first part of the UDI code), a detailed device description, and information about the labeler including the DUNS code, and company name and address.
- Commercial distribution - This includes the distribution status—whether or not the device is in commercial distribution, and the distribution end date—when the device will no longer be distributed.
- Alternative identifiers - If the device has another DI, either a direct marketing DI, a distinct packaging DI, one from another issuing agency, or one that was used previously, this information must be provided.
- Customer contact information - A phone and email address for patients or consumers who have questions about the device.
- FDA codes and listing number - If the device completed a pre-market authorization (PMA) that should be included as well.
- Manufacturing information - Manufacturing date, lot or batch number, serial number, and expiration date for the device.
- Latex information - Whether or not the device or its packaging contains rubber components.
- Device dimensions - What is the clinically relevant size and unit of measure for the device.
- Storage and handling - Requirements and parameters for storage including temperature, humidity, and pressure.
- Sterilization - Whether the device is packaged as sterile or requires sterilization prior to use.
The FDA provides a detailed spreadsheet of data requirements that you can use to prepare your submission.
Creating a GUDID submission
In addition to gathering the required information (and obtaining a UDI code for your device) there are several additional steps to complete in order to create a GUDID submission for your product. First, if you don’t have one, you’ll need to create a GUDID account. The FDA allows you to request an account online. Note that you will need to have a DUNS number for your business. If you don’t have one you can request one from Dun & Bradstreet at no cost.
There are two ways that you can enter your submission. You can do this online through the GUDID Web application. The FDA also allows you to submit your GUDID information all at once using an XML file that complies with Health Level 7 (HL7) Structured Product Labeling (SPL) formats. These submissions are made via the FDA Electronic Submissions Gateway, and require you to set up a gateway account.
In addition, some software providers (like Rimsys) include the ability to make electronic GUDID submission directly from their tools. They provide a system to organize and manage UDI data for the US and other countries, and can ensure that GUDID information for your products is kept up to date.
The global proliferation of UDI regulations
The GUDID was one of the first public databases of medical device information, but many additional countries and regions have followed suit. The European Union, China, South Korea, and Taiwan have all introduced UDI databases and requirements that manufacturers submit records for all of their products sold in-market, and ensure that they are kept up to date.
For more information about global UDI programs and timelines, see our UDI quick reference guide. And you can find more detailed information about the specific requirements in the EU and China in our Ultimate guide to the MDR/IVDR UDI and Ultimate Guide to the China NMPA UDI requirements ebooks.

Rimsys raises $16M Series A to bring regulatory order to the medtech industry
We’re excited to announce that we’ve closed $16M in Series A financing led by Bessemer Venture Partners, with participation from Allos Ventures, Private Opportunities, and Innovation Works. Rimsys was created because there wasn’t a viable regulatory information management (RIM) solution on the market for medtech companies, leaving regulatory affairs (RA) teams to manage increasingly complex work with spreadsheets. The growth that we’ve seen (3X this year), and the work we’ve done with some of the world’s largest medtech companies including Johnson & Johnson, Terumo, Siemens, and the Cooper Companies, makes us incredibly excited about what’s to come.
Regulatory digitization and automation for the medtech industry
The regulatory landscape for medical device, in vitro diagnostic, and medical software products is growing increasingly complex. The implementation of the new European Union Medical Device Regulations in May, to be followed by the In Vitro Diagnostic Regulations next year, brought new general safety and performance, unique device identification, and post-market surveillance requirements that manufacturers must comply with. Research from MedTech Europe predicts that as many as 76% of products will be withdrawn from the market as a result.
Growing complexity isn’t limited to the EU region. This year, Australia has released new essential principles requirements, Canada has expanded post-market requirements, and China has launched a new UDI system. The simple fact is that the largely manual way that RA teams have managed processes won’t work moving forward.
The Rimsys RIM Platform provides an automated, digital alternative to these traditional approaches. It’s a 100% cloud-based software solution that’s specifically designed around medtech regulatory activities and processes.
Rimsys provides a centralized "single source of truth" for all regulatory information and documents. It automates regulatory submissions, including product and UDI registrations, and monitors expiration dates, applicable standards, and regulations for changes that might impact products. It’s a single, integrated solution that supports the full breadth of regulatory activities, and organizes all of it at the individual product level, giving medtech companies unprecedented visibility into and control over their regulatory processes.
New leadership to drive continued growth
We’re also excited to announce new company leadership that will help us through our next phase of growth. We’ve added two new executive leaders with extensive industry experience: Adam Price, former head of post-market surveillance at Philips, and Christine Robertson, former IT leader supporting regulatory at Thermo Fisher Scientific. Adam will lead post-market strategy for Rimsys, developing new offerings to streamline and simplify that part of the regulatory lifecycle. Christine will lead implementation and professional services, bringing best-practices from successful large-scale RIM deployments to all of our customers.
We’ve also added two new board members: Andrew Hedin, Partner at Bessemer Ventures, and Eric Boduch, Co-founder of Pendo, a $2.6B SaaS company that helps companies develop better software products. Both bring a wealth of start-up, SaaS, and industry experience that will be incredibly valuable as we scale the company.
What’s next
Our goal is to build a comprehensive software platform for medtech regulatory affairs that supports activities across the regulatory lifecycle from pre-market to market placement to post-market surveillance. This year we became the first vendor to offer UDI management directly integrated with product registration data. And we announced a new partnership with Clarivate to bring world-class regulatory intelligence into the Rimsys platform where customers can leverage it directly within automated processes.
We will continue to expand our capabilities at an even faster pace, with new collaborative submission authoring, electronic transmission, regulatory intelligence, document management, and post-market surveillance features coming to the platform. We will also continue to expand our team with a new UK office to better serve the European market, and a number of new roles across the company. If you’re interested in helping medtech companies get lifesaving products to market more quickly, we’d love for you to join our team.
Learn more about Rimsys
We’d love to show you how the Rimsys RIM Platform can help medtech companies streamline processes across the regulatory lifecycle, strengthen global compliance, and get new products to market faster. Contact us to schedule a free custom demo.

RIM 101: what is regulatory information management?
Regulatory Information Management (RIM) refers to a category of software solutions that are designed to support and streamline the activities of regulatory affairs (RA) teams. For most teams they are a net-new category of software, and generally replace manual processes that are paper-based or run using traditional productivity software (spreadsheets and docs). RIM systems first emerged to support pharmaceutical regulatory activities, but in recent years medtech-focused solutions have hit the market as well.
Given their general new-ness, especially for medtech RA teams, it’s not surprising that many teams are unfamiliar with the technology. In our, admittedly informal, survey of RAPS 2021 attendees, only 11% of respondents said they currently use a RIM system, and 33% had no knowledge of the category at all. This article provides some background on what RIM systems are, and what they do to help medtech RA teams operate more effectively.

The role of regulatory affairs in medtech
To understand RIM systems, first we have to look at the role of regulatory affairs. In medtech, which includes medical devices, in vitro diagnostics, and medical software, RA teams play a critical role across a product’s lifecycle.
Before products are released for sale, RA teams work closely with research and development (R&D) teams to ensure that a new product meets necessary local requirements to be legally marketed in the desired target markets. There are over 113 different regulatory regimes around the world that medical devices are subject to. While there are many similarities, RA teams must understand the nuances between countries and guide R&D to ensure that products are developed accordingly.
Once products obtain market clearance, RA teams switch to monitoring mode to ensure that products can remain on the market. This includes keeping track of expiration dates and certificates, any changes in regulations or international standards that could impact the product, and any changes in the product or it’s technical documentation. Health authorities in many countries regularly perform product audits, so keeping all information in order and up-to-date is an important part of regulatory work.
RA teams usually take the lead on post-market surveillance activities as well, working closely with their quality assurance (QA) counterparts. They track adverse events and complaints, compiling this information from public and internal sources, and ensure that the data is reported appropriately to health authorities. Not all markets require extensive post-market surveillance for medical devices, but these regulations are becoming more common. Both the EU and Canada have recently implemented expanded surveillance requirements including the need for regular summary reporting to continuously confirm product performance and safety.
The information challenge
All of the regulatory activities highlighted in the previous section are repeated for every individual product the company sells in every regulated country or region. And, all of these activities are highly dependent on specific information. To do their jobs effectively, medtech regulatory affairs professionals need insight into global regulations and standards, detailed product specifications, testing, performance, and safety data, and a full record of all regulatory registrations and processes.
The problem is that this information is often scattered across the company. It’s stored in multiple systems, (sometimes physical) documents, and individual employees’ heads. Because this information is so scattered, RA professionals can spend up to 50% of their time just looking for things, and simple requests such as identifying whether a product has clearance to be marketed in a specific country can take days to complete.
How RIM systems can help
At a fundamental level, RIM systems are about helping RA teams corral and manage all of the information they need to do their jobs. RIM systems serve as a “single source of truth” for RA teams. They store and manage regulatory documents, integrate with systems across the company, and create a traceable record of all regulatory activities. All of this information is linked to individual products and countries or regions, making it much easier to find.
All of the collected information in a RIM system can be used to streamline regulatory activities across the product lifecycle. Before products are released, they provide access to regulatory intelligence, including market entrance requirements, that RA teams can use to guide product development and regulatory submissions. RIM systems also provide a collaborative digital hub where teams can author and assemble supporting documentation for new regulatory submissions.
For products currently on the market RIM systems can monitor registration expiration dates, and track changes in relevant standards and regulations to identify potential product impacts. This automated monitoring can give RA teams an “early warning”, and allow them to accommodate changes that might impact the selling status of a product.
RIM systems can also help with post-market surveillance activities. They can collect and centralize post-market data analytics, and facilitate planning and active surveillance activities to meet the most current regulatory requirements. These systems can also ensure that actions and conclusions drawn from the post-market surveillance process are consistently applied throughout the quality management system. And, the same authoring capabilities used to assemble pre-market submissions can be used for post-market reporting and communication with regional regulatory authorities.
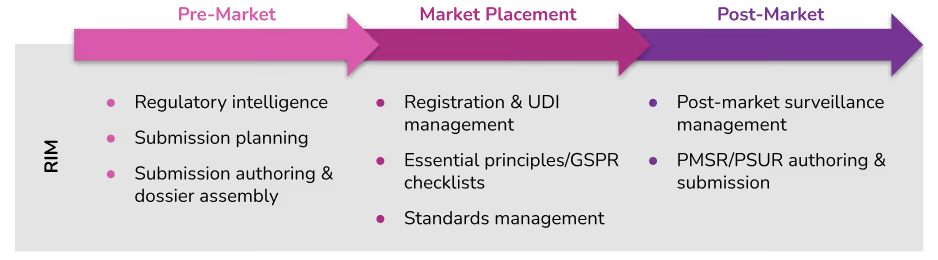
Project planning, tracking, and management
Underpinning all of these capabilities is a full set of project features that allow RA teams to effectively manage and track their activities. This can include project request features that allow internal teams or 3rd-party partners such as local distributors to request specific regulatory activities or information. RIM systems also provide project task management, approval workflows, and digital signature capabilities that are fully auditable, and 21 CFR Part 11 compliant.
RIM systems also provide detailed reporting in the form of customized dashboards and registration, product, standards, and documentation reports. These reports offer at a glance monitoring of key information and detailed visibility into regulatory status and activities. For many teams this level of visibility is new, and allows them to fully measure, benchmark and report on their activities to company leadership.
The impact of RIM systems
RIM systems can have a tremendous impact on RA teams. By centralizing information they improve team productivity by ensuring that up-to-date information is always easily available and consistently applied. By automating workflows like new submission creation, or essential principles/GSPR table assembly they ensure that work gets done quickly and in-line with country/region requirements. RIM systems also provide more visibility into regulatory activities, allowing teams to benchmark and more accurately forecast the time required for new market clearance, and other product milestones.
To the company, the increased regulatory efficiency and effectiveness means reduced revenue risk from noncompliance or having to pull products from market, stronger, more confident global regulatory compliance, and ability to get new products to market much more quickly.
To learn more about RIM systems, their key capabilities, and if your organization could benefit from bringing one onboard, read our RIM System Buyer’s Guide for Medtech Companies.

Regulatory information management (RIM) systems and organizational change management
At first glance, the juxtaposition of RIM and change management seems a little strange. One is a software tool and the other a management discipline, but one of the things we’ve seen across RIM deployments is that it’s difficult to have one without the other. For many regulatory affairs teams, a RIM system isn’t simply a tool, it’s a digital transformation. This means that there’s a broader set of organizational considerations and actions that need to surround the implementation of a RIM system to ensure its success. Remember that 70% of digital transformation initiatives fail.
RIM systems are a disruptive technology
Disruptive? Really? Aren’t RIM systems supposed to streamline regulatory activities, and improve team productivity? Yes they definitely provide these benefits, but they also require a change in how the team works. Most RIM implementations aren’t replacing existing software, they’re replacing manual processes. In our experience, Rimsys is displacing registrations that are managed via spreadsheets, and sometimes even paper-based processes.
This means that the way that teams have managed processes is changing significantly. While it’s likely that teams are struggling to operate effectively (there’s usually some organizational pain that leads to a RIM evaluation), there’s also discomfort with the change. RA team members are proficient in their work, they know how to get things done, and likely have systems they’ve created to cope with the inefficiencies in their current processes.
Regardless of department or industry, automation initiatives can lead to employees feeling threatened with obsolescence, lacking direction, and afraid of being replaced. In medtech regulatory affairs this is rarely the case. Most companies have to invest heavily in external consultants just to keep pace with the current workload. In fact, large medtech companies regularly outsource 50% or more of their regulatory activities. This doesn’t mean that team members won’t experience these insecurities. That’s why it’s important to have a change management strategy in place to support any RIM rollout.
A RIM change management strategy
All of these factors mean that RIM implementation that doesn’t have an accompanying change management strategy won’t see the same level of success, or deliver on expected outcomes. The good news is that there are a universal set of tactics to support effective change management that can be easily applied in this scenario. Here are 4 steps that you can take to lay the groundwork for a successful RIM implementation.
Step 1: start at the top
Teams that are in the process of acquiring a RIM system likely already have leadership support, but it’s important that your senior leaders have a visible presence in the process. This means issuing communications, participating in kick-off meetings, and being available to answer questions. This applies both to RA leaders and those in adjacent departments like QA and IT as well.
The visible support reinforces the idea that leadership teams are aligned and fully supportive of the changes taking place. It affirms that RIM is a strategic priority for the company, and helps to alleviate any fear or anxiety about the change. Leadership support also helps to signal to teams that they’ll be supported as they go through the implementation process, and that work will be prioritized.
Step 2: communicate early and often
RA teams are busy—often very busy. This is typically why a RIM system is being implemented in the first place. However, when teams are really busy, it’s really easy for communications to fall through the cracks. This means that plans and timelines for a RIM implementation need to be communicated more than once.
Communications should emanate from leadership teams (see step 1), and be candid about coming changes, the reasons for them, and the expectations from team members as the project moves forward. Leaders should encourage communication that moves in both directions, and be open to feedback from team members. Companies should look to create channels for RA team members to reach out with any comments or concerns.
Step 3: strive to minimize disruption
While there’s no way to completely eliminate the disruption associated with a new RIM system—it will fundamentally change the way a RA team works there’s no way around it—there are ways to minimize disruption. There first part of this is making sure you’re communicating enough about the project (see step 2). Team members are much more receptive to change if they aren’t blindsided by it.
It’s also helpful to take steps to make sure that team members have an opportunity to learn about the RIM systems throughout the acquisition and implementation process. Bring team members into product demonstrations, and let them ask questions about solutions that are being evaluated. Don’t wait to run training sessions until the RIM system is fully implemented. These can run in parallel. With this approach the whole team feels invested in the solution, and is fully ramped to start running at the end of implementation.
Step 4: lay the groundwork for continuous improvement
This article discusses change management from the perspective of a discrete event—the acquisition and implementation of a RIM system. However, RA teams shouldn’t look at change as something with fixed start and end, but rather as something continuous. RIM systems today represent one way that RA teams can embrace digitization and automation to improve how they work. There will be many additional opportunities as regulations, regulatory bodies, and technology evolve.
In recent years we’ve seen an expansion of UDI requirements for medical devices across markets. We’ve seen more stringent requirements for post-market surveillance. And we’ve seen the growing adoption of digital pathways for regulatory submissions and other interactions with health authorities. For many teams, a successful RIM implementation is just the first step on what will be a broader organizational transformation.
Successful changes for RIM and beyond
RIM systems can provide enormous benefits to RA teams, but only if they’re fully implemented and adopted. While deep in the weeds of requirements gathering and vendor evaluations it’s easy to overlook the fact that moving from traditionally manual processes to automated ones in a RIM system represents a significant organizational change. As a part of any RIM acquisition initiative, teams should fully understand their change management needs, and take steps to address them in concert with software selection.
Having teams fully onboard and supportive of the initiative makes them much more likely to adopt the final solution. Engaging in this way also allows you to plan for, and prioritize the resources needed for the implementation phase—leading to faster time to value for the project. Ultimately organizational change will and should be something that RA teams are comfortable with. The practices adopted with a RIM implementation can be used to support future digitization and automation across all RA activities.
To learn more best practices around RIM sourcing and implementation including an organizational self-assessment, detailed overview of capabilities, and a worksheet you can use for vendor evaluations, check out our RIM Buyer’s Guide for MedTech Companies.

A primer on medical device classification
While it may seem simple, medical device classification can be a challenging task for many medical device and IVD manufacturers. Device classes for specific regions and countries have a number of small variations, and each of those variations can impact the process by which a device obtains market clearance. Getting it wrong can lead to delays in getting to market. This article explores the classification systems for three major markets, and their associated regulations.
An important component of achieving regulatory approval is the appropriate classification of a medical device or in vitro diagnostic device, according to the specific regulations within a country or region. Product classifications are related to the intended use of the product and the perceived risk that it poses to a patient using the device. While this general approach is pretty standard across all regions, there are many small variations in different country classification systems that can impact how a device is regulated. It would be much easier if there was one global classification system that everyone followed.
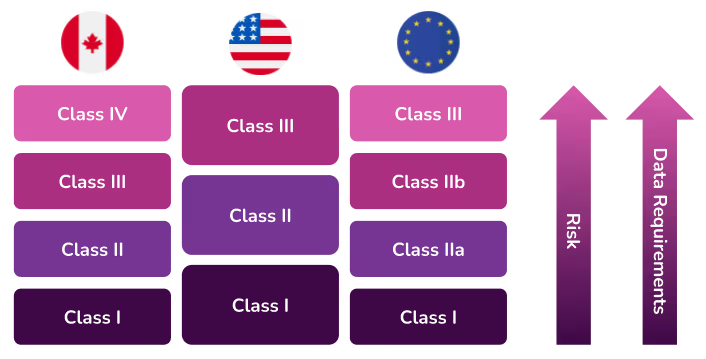
However, since there are different guidelines to classifying a medical device (per country), we’ll dig into the most popular classification systems—Canada, the European Union and the United States. These three are globally perceived to have strong, thorough, and trusted quality and regulatory systems. Their approaches are often mirrored or used as proxies for market clearance in other countries.
Canada
The Canadian Medical Devices Regulations include guidelines that classify devices into four risk classes. If a medical device can be classified into more than one class, the class representing the higher risk always applies.
- Class I devices do not require a medical device licence to be sold in Canada, but manufacturers, distributors and importers of these devices are required to obtain an establishment licence.
- Class II requires a medical device licence
- Class III requires a medical device licence
- Class IV requires a medical device licence
Some examples of different classes of devices in Canada include:
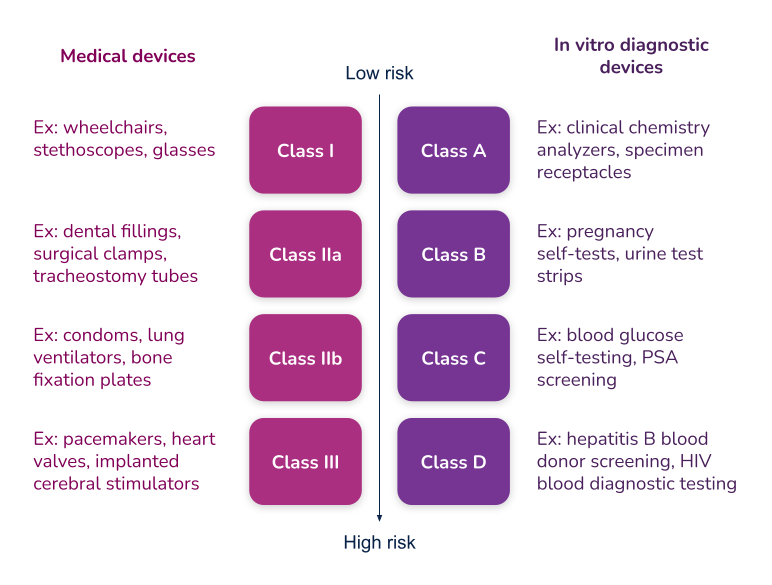
European Union
One of the main changes introduced with the new MDR/IVDR regulations are new classification rules for medical devices and in vitro diagnostic devices. If you have gone through the process of getting your medical device in the European market before, you might find it more difficult with the new EU MDR rules. For example, a new medical device you want to bring to market might now fall into a higher classification than it would have previously (under the MDD), and therefore require more testing, updates to documents, quality approvals, etc.
The new EU MDR brings the classification of medical devices in Europe more in line with international regulations, specifically the United States. These updated rules are listed in MDR 2017/745 for devices and IVDR 2017/746 for in vitro diagnostic products. As with Canada, if a medical device can be classified into more than one class, the class representing the higher risk always applies.
The EU has recently released a new guidance document MCDG 2021-24 to assist device manufacturers with device classification questions.
Class I – this classification is for the lowest risk device. Most medical devices in this category do not require a conformity assessment from a Notified Body so instead, they can be self-assessed. However, manufacturers must still complete a Technical File as part of the approval process.According to MDCG 2019-15, there are three subclasses under Class I. Unlike most Class I devices, these will require the involvement of a Notified Body.
- Class Im: a product with a measuring function
- Class Is: a product that is sterile
- Class Ir: a product that is a reusable, surgical instrument
Class IIa – this classification is for a medium risk device. A conformity assessment by a Notified Body is required for this classification.
Class IIb – this classification is for medium-to-high risk devices. A conformity assessment by a Notified Body is required for this classification.
Class III – this classification is for the highest risk devices. A conformity assessment by a Notified Body is required for this classification.
Examples of different classes of devices in the European Union include:
In Vitro Diagnostic Devices:
Class A – this classification is for the lowest risk in vitro diagnostic devices. Most IVD devices in this category do not require a conformity assessment. Instead, they can be self-assessed.
Class B – this classification is for medium risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Class C – this classification is for medium-to-high risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Class D – this classification is for the highest risk in vitro diagnostic devices. A conformity assessment by a Notified Body is required for this classification.
Examples of the different in vitro diagnostic device classes in the European Union include:
United States
In the United States, the Food and Drug Administration (FDA) is responsible for overseeing the safety of medical devices. The FDA has established classifications for approximately 1,700 different types of devices and grouped them into 16 medical specialties referred to as panels. All three classes of devices are subject to General Controls which are the baseline requirements of the Food, Drug and Cosmetic (FD&C) Act.
- Class I – General Controls (with or without exemptions)
- Class II – General Controls and Special Controls (with or without exemptions)
- Class III – General Controls, Special Controls and Premarket Approval
You are permitted to classify your own medical device based upon the FDA guidance documents and set regulations. However, if you wish for the FDA to assist with establishing your classification you can submit a 513(g) Request for Information. Note that there is a user fee associated with a 513(g) Request.
The device class determines which type of premarketing submission/application is required for market clearance.
In some instances, you do have the opportunity to reclassify your product after it’s been released to the market. The regulatory class of a device type, as defined by the Federal Food, Drug and Cosmetic Act (FD&C Act), may be changed through petition to the FDA. This process is only applied to a device type though, not to an individual device.
Examples of medical device classification in the US include:
Getting classification correct
Medical device classification is simple in that each country and region generally follows the same classification approach, and complex in that minor differences can change how a device is classified across markets. Understanding how a device is classified is one of the critical first steps regulatory affairs teams need to take when entering a new market, as medical device class often determines the pathway to market.
For example, in the EU classification can mean the difference between self-certification and a required conformity assessment from a Notified Body. In the US, classification can mean the difference between a 510(k) or PMA process for market clearance. Getting classification correct can ensure a smoother and faster route to market.
To learn more about market clearance processes for medical devices in the US, check out the Beginner’s Guide to the 510(k).

MDSAP - the ultimate guide to the medical device single audit program
This article is an excerpt from The ultimate guide to the medical device single audit program (MDSAP) ebook.
Table of contents
- What is MDSAP?
- History of MDSAP
- Who is responsible for the MDSAP?
- How does an MDSAP audit work?
- Audit sequence
- You got a nonconformity – now what?
- What does an MDSAP audit cost?
- Why choose the MDSAP certification process?
- Potential disadvantages of the MDSAP
- Ready to participate? – Here’s how to get started
- Completing a successful MDSAP audit
The Medical Device Single Audit Program (MDSAP) was designed and developed to allow a single audit of a medical device manufacturer to be applied to all country markets whose regulatory authorities are members of the program. The MDSAP provides efficient and thorough coverage of the standard requirements for medical device manufacturer quality management systems, and requirements for regulatory purposes (ISO 13485:2016). In addition, there are specific requirements of each medical device regulatory authority participating in the MDSAP that must be met:
- Conformity Assessment Procedures of the Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3)
- Brazilian Good Manufacturing Practices (RDC ANVISA 16)
- Medical Device Regulations of Health Canada (ISO 13485:2003)
- Japan Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No 169)
- Quality System Regulation (21 CFR Part 820), and specific requirements of medical device regulatory authorities participating in the MDSAP program.
This means that a report from a single MDSAP audit of a medical device manufacturer would be accepted as a substitute for routine inspections by all the member Regulatory Authorities (RAs) across the world. There are currently five participating Regulatory Authorities (RA) representing the following countries: Australia, Brazil, Canada, Japan and the USA.

In April, 2021, the RAs released an “Audit Approach” document (MDSAP AU P0002.006) that combines the formerly separate MDSAP Audit Model and Process Companion documents into a single guidance document. It includes guidance for assessing the conformity of each process and includes an audit sequence, instructions for auditing each specific process, and identifies links that highlight the interactions between the processes.
In March 2012 the US FDA announced that they had approved a final pilot guidance document “Guidance for Industry, Third Parties and Food and Drug Administration Staff: Medical Device ISO 13485:2003 Voluntary Audit Report Submission Pilot Program.” This allowed the owner or operator of a medical device manufacturing facility to be removed from FDA’s routine inspection work plan for 1 year upon completing a ISO 13485:2003 audit. This guidance document went into effect in June 2012, and was intended as an interim measure while a single audit program was being developed.
This pilot program was not very successful and few companies signed up because they did not see any advantage in participating. The manufacturer had to pay for a third party to inspect their facilities, generate a report, and share the inspection results back to the FDA. Many companies were reluctant to contract “someone else” to perform their inspection when they could easily wait for the FDA to conduct an inspection for free.
During its inaugural meeting in Singapore in 2012, the International Medical Device Regulators Forum (IMDRF) appointed a working group to develop a set of documents for a harmonized third-party auditor system. Hence, the “Medical Device Single Audit Program” (MDSAP) was formed. The concept was similar to the FDA’s original idea of creating a third-party auditor to help reduce their workload of performing regulatory audits of medical device manufacturers’ quality management systems. This new approach would consist of a single audit that would review regulatory QMS compliance, conducted by a third-party, who would later be called an Auditing Organization (AO).
From January 2014 to December 2016, five countries participated in a Medical Device Single Audit Program Pilot. In June 2017, a report was generated summarizing the outcomes of prospective “proof- of-concept” criteria established to confirm the success of the program. The outcomes are documented in the final MDSAP Pilot Report and recommended that the program become fully active and open to any manufacturer who requested this type of audit.
The governing body of the MDSAP is the Regulatory Authority Council (RAC), which is composed of two senior managers (and a few other staff members) from each participating RA. They are responsible for executive planning, strategic priorities, setting policy, and making decisions on behalf of the MDSAP International Consortium. The RAC also reviews and approves documents, procedures, work instructions, and more. The mission of the MDSAP International Consortium is to jointly leverage regulatory resources to manage an efficient, effective, and sustainable single audit program focused on the oversight of medical device manufacturers on a global scale.
Other international partners that are involved in the MDSAP include:
MDSAP Observers:
- European Union (EU)
- United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA)
- The World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Program
MDSAP Affiliate Members:
- Argentina’s National Administration of Drugs, Foods and Medical Devices (ANMAT)
- Republic of Korea’s Ministry of Food and Drug Safety
- Singapore’s Health Sciences Authority (HSA)
The observers and affiliate members are not the same as the participating member RA’s. The observers simply observe and/or contribute to RAC activities. Affiliate members, on the other hand, are interested in engaging in the MDSAP program and are subject to certain rules. They are only given access to a certain level of information about the manufacturers, audit dates, and information in audit reports.
They are also invited to attend sessions that are open to members, observers, and affiliates only.
Audits can also be conducted by MDSAP participating RAs at any time and for various reasons including:
- "For Cause" due to information obtained by the regulatory authority
- as a follow up to findings from a previous audit
- to confirm the effective implementation of the MDSAP requirements
The purpose of audits conducted by the RAs is to ensure appropriate oversight of the AOs MDSAP auditing activities. The AOs are appointed by the RAs and a list of the currently approved AO’s is published on the FDA website. Most AOs offer a broad range of management system certification services, beyond just medical devices. Manufacturers should verify that prospective AOs are clearly trained and perform MDSAP audits of medical devices.
AOs have the final word as to whether a manufacturer has met the requirements for the MDSAP during the execution of the audit and generation of the associated reports summarizing the results. MSDAP RAC participating RAs have the final decision regarding all development, implementation, maintenance, and expansion activities associated with the program.
Although an unannounced visit by an AO is rare, it can happen in circumstances where high-grade nonconformities have been detected.
To continue reading this eBook including a detailed look at the MDSAP audit process and grading, pros and cons of the approach, and how to get started please register to download the full version.
