


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The beginner's guide to the FDA De Novo classification process
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
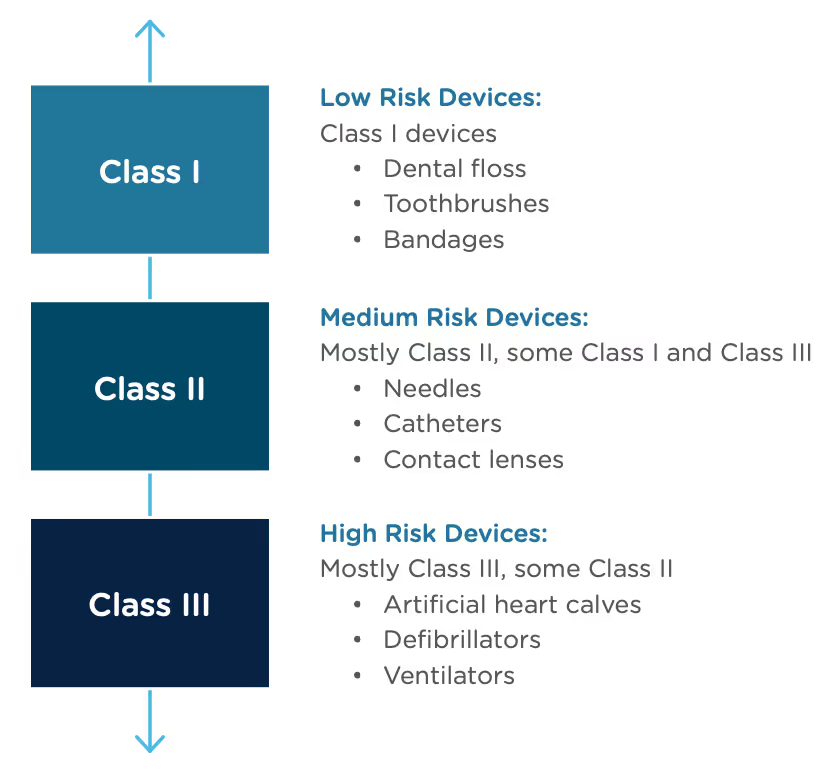
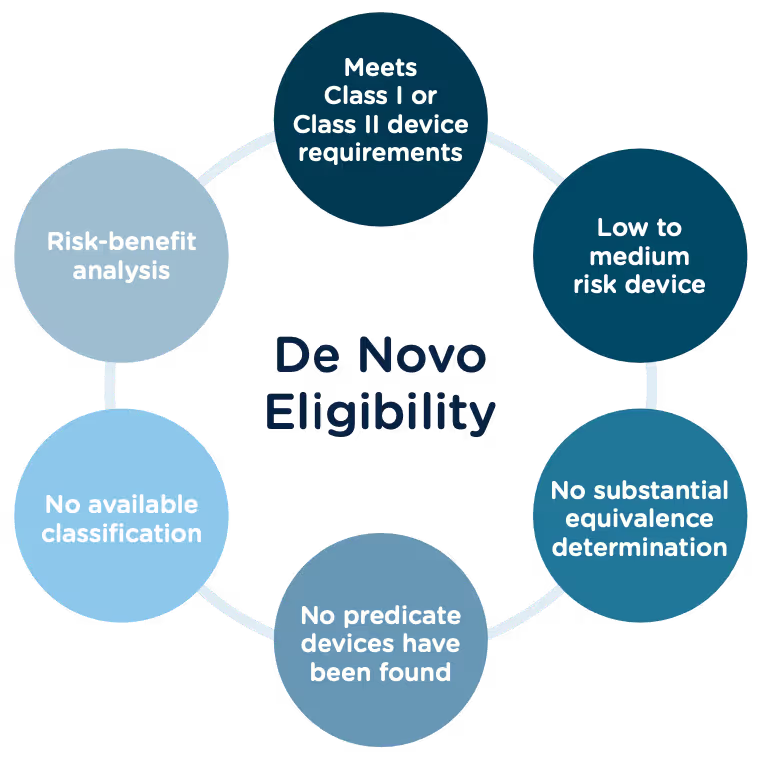
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.




Dispatches from RAPS Convergence: The state of regulatory tools
A few weeks ago we attended (virtually) the RAPS Euro Convergence conference. The event, despite the virtual format, still brought together regulatory professionals from across the European region for several days of immersive learning. At Rimsys, we took advantage of the opportunity to explore the state of regulatory tools and processes in the region, and see how they compare to those of North American teams.
Visitors to the Rimsys booth, both at RAPS Euro Convergence and at last year’s North American RAPS Convergence event were invited to fill out a short survey in exchange for the opportunity to win an Amazon gift card. While the respondents were self-selected, and the results aren’t statistically significant, they still showcase some interesting differences in the tools and mindset of regulatory affairs professionals in each region.
Regulatory tools used
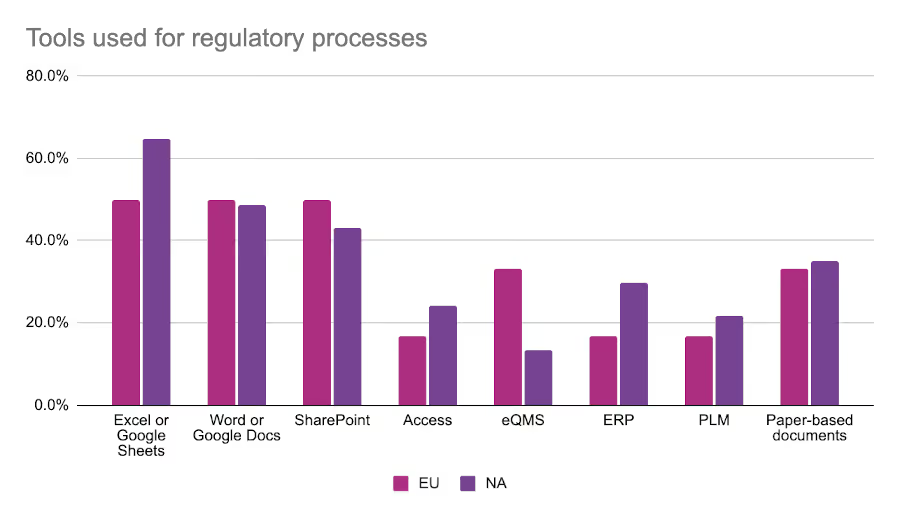
Across both regions, Excel is the most commonly used tool by regulatory teams. Over half of respondents across both regions reported using Excel to manage regulatory information and processes. European regulatory affairs professionals were much more likely to use an electronic quality management system (eQMS) to manage their work, and about one-third of respondents to both surveys indicated that they used physical paper-based documents.
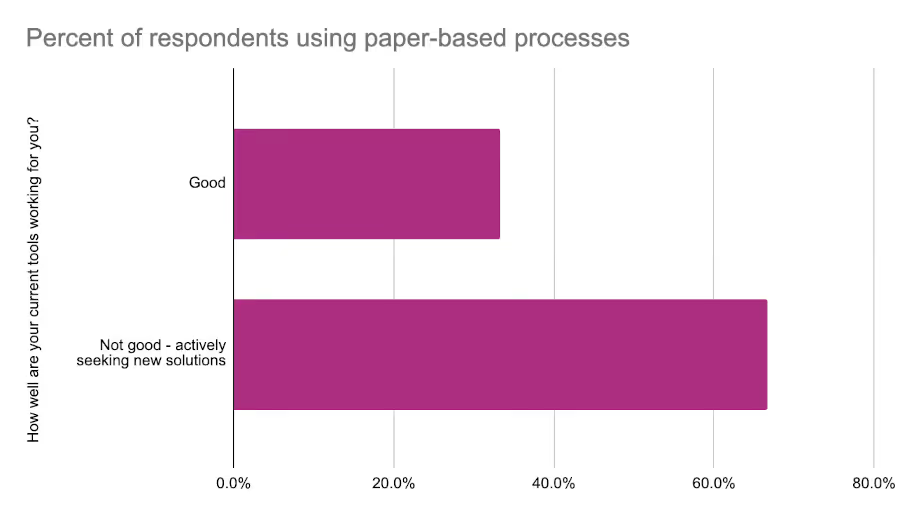
We also asked respondents how well their tools were working. European respondents were generally more content with their toolset with 66% saying their tools were “good”. By comparison only 22% of North American respondents felt the same. One thing that was clear was the impact of paper-based processes on satisfaction. Respondents who reported struggling with their current tools were nearly twice as likely to use paper-based processes as part of their regulatory activities.
Work efficiency and satisfaction
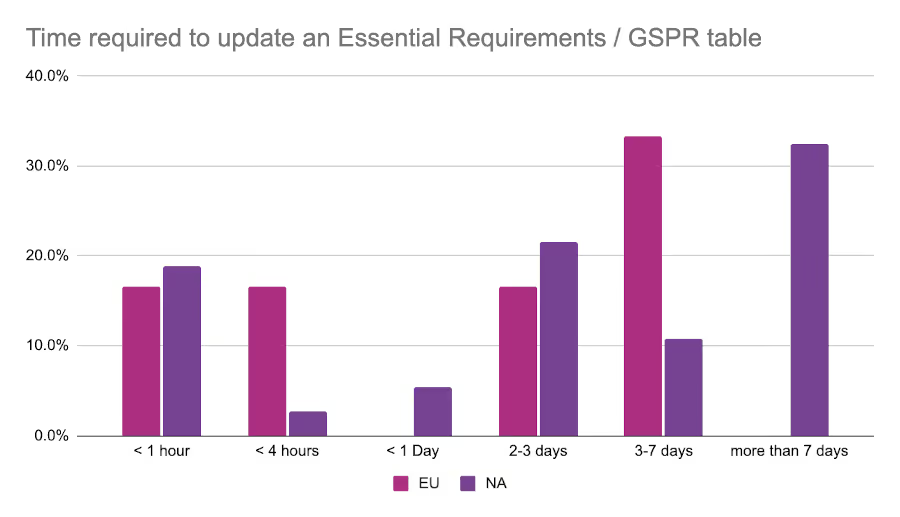
Just over 50% of all respondents indicated that the tools they use “could be better”. This may have something to do with the amount of manual work that RA teams find themselves doing. As part of the survey we asked respondents how many hours it takes on average for them to update one of their Essential Principles / General Safety and Performance Requirements (GSPR) tables. The most common response from the EU event was 3-7 days, while those at the North American event were most likely to report greater than 7 days for the same task.
Given their slightly better estimated performance when it comes to regulatory processes, respondents from the EU were less likely to express frustration with their roles and dissatisfaction with their productivity. We asked attendees at both conferences to rate their “regulatory frustration” and satisfaction with their productivity on a scale from 1 to 5 with 1 being very frustrated/unsatisfied, and 5 being very satisfied/not frustrated at all.
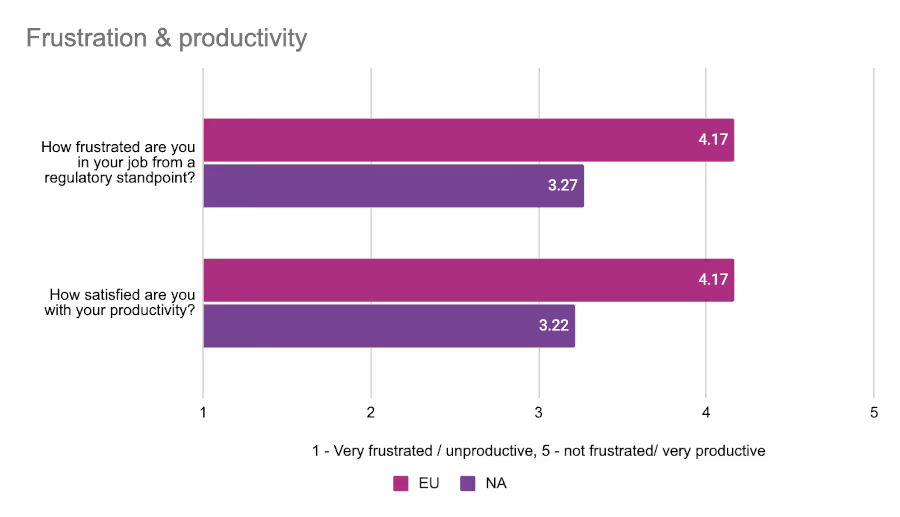
EU respondents were noticeably more positive in their assessments than our North American respondents, although everyone generally had a favorable view of their productivity.
Implications
Looking at the survey results, there are some interesting discrepancies. Teams in both regions heavily use poorly-suited tools and manual approaches to regulatory processes, yet have generally favorable views of their productivity. This points to two likely conclusions. One, that regulatory affairs professionals are particularly comfortable with a lot of manual administrative work, and two, that they’re largely unaware of the new class of regulatory information management (RIM) solutions that are specifically designed to automate and streamline regulatory workflows.
RIM platforms (like Rimsys) provide a digitized central repository for regulatory information and content, allowing RA professionals to streamline product submission, authorization, and maintenance processes (such as updating GSPR tables that we discussed earlier). RIM platforms can reduce the time and resources required to complete regulatory activities by more than 50%.

Building a business case for a RIM system
While the space is growing quickly, regulatory information management (RIM) systems are still relatively new to a lot of medtech companies. RIM systems help companies digitize and automate regulatory activities associated with their products. They provide a central information repository for all regulatory content and streamline activities like market registrations, data capture and transmission of unique device identification (UDI), and building essential principles tables while tracking associated standards.
Given that many regulatory affairs teams still manually manage these processes through complex spreadsheets and disjointed documents, the value of an automated solution is pretty obvious, but quantifying that value—especially for teams that aren’t seasoned software acquirers—can be a bit harder. This guide provides a framework for regulatory affairs teams to quantify the potential benefits of a RIM system, and build an internal business case for investment.
Challenges with the traditional approach to regulatory affairs
While functional, there are a number of painful inefficiencies that come along with traditional approaches to regulatory processes. Without an automated way to keep track of all the information and supporting documents associated with these processes, companies struggle with:
- Lack of visibility into regulatory data, clearance/approval status for different markets, and time-to-market metrics
- Compliance gaps driven by disconnects between go-to-market, distribution, and regulatory affairs teams
- Difficulty assessing the impact of, and responding to changes in standards or regulations
- Accumulation of “tribal” knowledge among individuals that limits continuity and visibility across the organization
These pains represent specific costs to MedTech companies in the form of:
- Staffing: Companies must over-staff regulatory affairs teams to support highly-manual processes. Expensive consultants are often brought in to help address in-house resource shortages.
- Productivity: Regulatory affairs teams lose huge amounts of time repeatedly hunting for information (up to 50% of their time spent).
- Lost revenue: Long application times and lack of process visibility delay market entry for new products. Non-compliance can lead to fines, or the need to pull products out of specific markets.
The automation and data consolidation/integration provided by a RIM system can significantly reduce these costs, and provide a clear, measurable return on investment.
Additional benefits of a RIM system
In addition to the addressing the pains outlined above, RIM systems can provide valuable benefits across MedTech companies:
- IT teams: Without a bespoke platform to manage regulatory processes, regulatory affairs teams rely on a broad collection of tools to support their day-to-day work. This can include specific software to create and manage UDIs and access regulatory intelligence, as well as use of software designed for other functions: enterprise resource planning (ERP), product lifecycle management (PLM), or quality management systems (QMS)—highly-configured to try and support regulatory activities. A comprehensive RIM system (like Rimsys) provides support for multiple regulatory functions, saving IT teams the cost of acquiring and maintaining separate systems. With functionality specifically designed for regulatory processes, a RIM system is easier to support than customizations to tools designed for other functions.
- Go-to-market teams: Sales and marketing organizations can also benefit from the adoption of a RIM system. In addition to bringing new solutions to market more quickly, RIM systems can also help with planning and forecasting. Visibility into the time and cost required to enter different markets, and the specific regulations associated with each market, can help go-to-market teams better prioritize target markets, and set revenue projections for their product lines. RIM systems can also provide workflows for project requests, allowing go-to-market teams to better coordinate registrations to support planned product launches.
- Distributors, in-country sponsors, and notified bodies: One-off email communications with external parties is not only time consuming for regulatory affairs teams, but inefficient for partners. RIM systems can provide controlled access for external parties, allowing them to login and directly access needed information without sending an email, and waiting (sometimes days) for a response. Streamlining these communications allows partner organizations to move more quickly, and ultimately accelerate the delivery of products into new markets. This helps to grow revenue while also improving the productivity of regulatory affairs teams who no longer have to interrupt their work for every internal or external information request.
Building your business case
Putting together the challenges and benefits outlined in the previous section (as applicable to your team), will feed the primary content of your business case. Next we’ll take a look at how to structure the content.
Part 1 – Your current situation and challenges
One of the best places to start when putting together a business case for a RIM system is to look at your current situation. What are the challenges that your organization faces? How much time is spent looking for information, submitting marketing applications, completing regulatory impact assessments, creating and maintaining the new MDR / IVDR GSPRs? How long does it take your team to complete new registrations? How well do internal teams communicate and coordinate go-to-market activities for new products and markets? How many products and countries does your team support today? What would happen if that number increased significantly?
Next, look at the business implications of the current situation. Does your team have a backlog of requests from go-to-market or other management teams? How often do you bring in external consultants to help with workload? Note the associated costs of your project backlog (delays in receiving market clearance), and the amount spent each year on consultants. Are there any other business risks such as continuity or non-compliance that are associated with your current approach? Does your team have the ability to identify regulatory roadblocks associated with markets you’re planning to enter?
Part 2 – Recommended solution
Here’s where you make the primary case for a RIM system. Highlight the specific capabilities and benefits of your solution of choice, and how it will address the challenges of your current situation. Highlight areas where other teams will benefit from the solution, and opportunities to drive greater organizational alignment.
Part 3 – Costs and expected ROI
Estimating costs and returns can be difficult, but it’s one of the key ways to improve the credibility of your business case. If you’re unsure about costs you can generally assume that a full-featured RIM system will cost the equivalent of 1 to 3 full-time regulatory affairs professionals, and use that as a starting point.
When calculating returns start with time-savings. If regulatory activities could be completed 50% more efficiently, how would that impact the number of consulting hours you use or hiring plans? If you could reduce pre-market clearance time by 6 weeks, how much additional revenue could be generated? Have you had instances where you incurred fines or had to remove products from a market due to compliance issues (i.e. shipping to markets where the product hasn’t been approved)? What were the associated costs?
Next, look at what your RIM system is replacing. Do you have individual tools for UDI or other functions that could be replaced? What about other tools? Does your team have seats in ERP or QMS tools that won’t be needed any more? All of these are relevant cost savings. In most cases, a RIM system will show strong ROI once you’ve estimated all of these savings.
Part 4 – Alternatives
Here’s where you want to demonstrate that you’ve done your due diligence in support of your recommendations. Have you evaluated multiple software solutions? Enumerate the specific capabilities or approach that led you to recommend your preferred vendor. Are there other ways to address your current challenges: expanding the regulatory affairs team, or implementing other types of software (rather than a RIM system)? Explain why you think these solutions will be less effective.
Putting it together
RIM systems can provide significant benefits specifically to regulatory affairs teams and broadly across MedTech companies. With a bit of legwork, it’s easy to put together a compelling case for investment in a solution. What can sometimes lead to difficulty in quantifying potential benefits—the fact that many MedTech companies don’t have a robust measurement framework for Regulatory affairs processes—means that you’ll be in a much better position to assess the performance of your team and impact of your investment once the analysis is complete.
Ready to get started? Download our RIM business case template. Questions? Our team is happy to work through the process with you and validate your estimates based on the experiences of our clients.

5 ways a RIM system can accelerate time-to-market for MedTech companies
Like all products, time-to-market is a critical success factor for medical technology (medtech). Product research and development ties up capital investment that can only be recouped when products start selling. For start-up and early stage companies, time-to-market can be the difference between success and failure. With a limited capital runway, they must demonstrate market success to access additional funding and growth opportunities. Unlike other products, however, medical devices have an added hurdle of regulatory clearance that must be obtained before products can be marketed and sold.
Underestimating the regulatory burden
It’s easy to underestimate the amount of time and effort required for regulatory activities related to New Product Introductions (NPI), especially during the development process. Policies, procedures, and submission processes can vary significantly between countries and regions, and almost all of them require government clearance before devices can be marketed. For larger companies it’s not unusual to operate in 100+ countries, creating an enormous array of standards and applications that must be manually managed.
These challenges are exacerbated by unclear lines of communication and siloed information across systems. Product development and testing information is stored within Product Lifecycle Management (PLM) and Quality Management System (QMS) solutions. Sales forecasting, marketing, and production information is stored within Enterprise Resource Planning (ERP) and Customer Relationship Management (CRM) systems. And regulatory, standards information, and compliance documents are stored across various file systems.
Regulatory submissions must synthesize information from R&D, QA, and go-to-market teams, and data from across all of these systems. Any mis-steps, or missing information can result in significant go-to-market delays, and even rejections from regulatory bodies. Coordinating regulatory status between go-to-market regulatory affairs teams can be a particularly vexing challenge. Without clarity about what markets have been cleared/approved, companies run the risk of moving too quickly ahead of the regulatory process, or unnecessarily delaying market entry.
What is a RIM system?
Despite all of the complexity associated with regulatory submissions, many teams rely on rudimentary approaches to coordinate and manage them. Submissions and associated data are managed via individual documents or complex color-coded spreadsheets, with no central repository of submission records or status.
Regulatory Information Management (RIM) systems are software solutions designed specifically to help companies streamline the submission, authorization, and maintenance process. They provide a digitized central repository for all regulatory information and content, allowing companies to automate and maintain compliance in the global market.
Using a RIM system to manage regulatory submissions and compliance can improve efficiency and productivity, reduce the risk of rejected submissions and noncompliance, and provide greater visibility into ongoing registration processes and status across the organization. While RIM systems provide specific benefits to regulatory affairs teams, they also provide company-wide benefits—specifically when it comes to accelerating time-to-market for new products.
5 ways a RIM system can accelerate time-to-market for new products
Full-featured RIM systems (like Rimsys) provide a number of capabilities that MedTech companies can take advantage of to get new products to market more quickly—and keep them there.
- Regulatory intelligence. Medical device registration requirements and standards vary across regions and countries. Understanding market entry requirements and timelines for regulatory submissions are necessary for any go-to-market planning, but finding and keeping track of this information can be challenging—especially for early-stage companies. RIM systems can provide up-to-date information about regulatory requirements without a lot of manual research and document management. Go-to-market teams can leverage the breadth of regulatory information in the system to develop rollout strategies for different markets, and identify the most attractive markets based on size and regulatory complexity.
- Digital forms and templates. Each regulatory submission requires multiple templates and forms, many of which aren’t available digitally. Finding the correct, current form, and manually filling it for each country/region can add significant time to the regulatory submission process. RIM systems can take the guesswork out of finding forms, by providing a library of digital templates for different markets that companies can easily access and fill. Even offline forms can be loaded into the system for digital filing and storage. With a modern RIM system, regulatory teams can easily access, fill, and track progress for submission forms in all the markets they’re looking to enter.
- Centralized visibility and information storage. RIM systems can function as a “digital hub” and single source of truth for all of the information associated with the regulatory process. Submissions require detailed product information, testing results, labeling, and other information that is often stored in other systems. Rather than sourcing this information over and over again, RIM systems provide regulatory teams with an organized repository that they can reuse across global applications. RIM access can be extended to other teams, and even external partners (like in-country distributors) to provide visibility into regulatory information and the status of submissions, making it easier to drive alignment around the process and coordinate go-to-market plans.
- Process integration across systems. Market authorization is one of the most critical pieces of information that go-to-market teams need. Maintaining that information across multiple products and multiple markets, and keeping it visible to go-to-market teams is a consistent challenge. RIM systems can integrate directly with ERP or CRM systems to feed authorization information directly into sales and distribution processes. Automating the in-country authorizations across systems can prevent noncompliance, and ensure that go-to-market teams are able to launch as soon as authorization is obtained. The same integration capabilities can be used to automatically retrieve product information from PLM and QMS systems, further speeding the application process.
- Automated regulation and standards tracking. This doesn’t necessarily directly impact time-to-market for new products, but it can definitely impact time in the market. Regulations and standards aren’t static, and regulatory affairs teams must keep on top of pending changes to ensure that products remain compliant and retain selling authorization. RIM systems can help to track changes, and flag products for potential compliance issues or that are at risk of losing authorization.In addition to regulatory changes, RIM systems can track authorization expirations, and other important events, helping companies maximize the revenue potential of their products by avoiding regulatory disruptions.
Accelerating regulatory approval and product go-to-market
MedTech companies are keenly aware of the role regulation plays in getting new products to market, but they aren’t always aware of the time and effort required to reach all of their target markets. Manual processes, disjointed information, and lack of coordination and visibility across teams can make it hard to obtain marketing authorization in a timely manner. This can significantly impact time-to-market—delaying return on investment, and even putting companies, themselves at risk.
RIM systems can eliminate a lot of the inefficiencies that slow down regulatory processes. By providing insight into regulatory requirements, access to digital templates, and integration across tools, they make it easier for companies to complete timely, successful regulatory submissions, and accelerate time-to-market.
RIM software from Rimsys
Rimsys is the only holistic RIM software designed specifically for medical technology companies. It helps companies digitize regulatory management by bringing together global UDI requirements, Essential Principles/GSPR, and regulatory registrations while monitoring products at the SKU level.
To learn more about RIM software from Rimsys, read our benefits datasheet.

Arena Solutions and Rimsys announce partnership to offer an end-to-end quality and product-centric regulatory solution
Foster City, Calif., January 12, 2021 – Arena Solutions, a leader providing cloud-based product development solutions for high tech, consumer electronics, and medical device industries, today announced a new partnership with Rimsys Inc., a world-leading provider of a holistic Regulatory Information Management (RIM) software platform designed specifically for medical technology (medtech) companies.
The Arena and Rimsys partnership offers a secure cloud-based, product-centric regulatory solution for the medtech industry. Rimsys seamlessly integrates with Arena’s QMS and PLM solutions by pulling product and documentation information directly into Rimsys to create, manage, and maintain marketing applications such as 510(k), Summary Technical Documentation (STED), and Table of Contents (ToCs).
The Arena product development platform connects product and quality processes allowing dispersed teams throughout the product design and manufacturing process to work together. Rimsys integrates with Arena’s platform by syncing product information so companies can better manage global registrations and selling status at the SKU level. Rimsys has the capability to pull in QMS records and documentation to create and compile regulatory applications from approved documentation. The integration automatically monitors for documentation changes and alerts users when updates occur with additional reporting based on document location ensuring a single and accurate source of truth.
"The seamless and deep integration between Rimsys and Arena solutions reduces the day-to-day regulatory management," said James Gianoutsos, Founder and President of Rimsys. "The administrative burden of compiling marketing applications and the maintenance of product data is completely eliminated, allowing for increased compliance, efficiency, and visibility throughout the organization."
"Our partnership with Rimsys makes it easier for MedTech companies to address regulatory affairs, product registration, and standards management more effectively," said George Lewis, VP of Business Development and Strategy for Arena Solutions. "This new integration streamlines regulatory compliance processes by accelerating the notification of updates to critical quality records and documents."
About Arena Solutions
Arena Solutions helps innovative electronic high tech and medical device companies create products that change the world. Arena unifies product lifecycle (PLM) and quality management (QMS) processes, allowing every participant throughout the product realization process from design to manufacturing to work together. With Arena, teams accelerate product development and delivery to increase profits. For more information, visit ArenaSolutions.com.
About Rimsys
Rimsys is a world-leading provider of Regulatory Information Management (RIM) software for medical technology companies. Built by and for regulatory affairs professionals, Rimsys digitizes, automates, and creates regulatory order to ensure products adhere to changing global regulations. It is the only holistic RIM software for medical devices, in-vitro diagnostics, and medical device software that makes it easy to manage global UDI requirements and navigate the pillars of regulatory affairs, including product registration, standards management, essential principles/GSPR, and regulatory intelligence. rimsys.io

Rimsys releases new automated unique device identification (UDI) module for its holistic medtech RIM platform
Rimsys, a world-leading Regulatory Information Management (RIM) software platform for medical technology companies, announced the release of an innovative and automated solution to address the global Unique Device Identification (UDI) requirements. This enhancement to the Rimsys ecosystem is immediately available to new and existing customers, providing a compliant method for tracking and traceability of UDI data that is fully compatible with global health authority databases.
UDI is a global harmonization effort to provide more consistent data that benefits overall product traceability throughout the medical device supply chain. It aims to adequately identify medical devices from manufacturing through distribution to patient use. Because global markets have different UDI requirements, product data can become a maintenance nightmare, even for the most mature medical technology companies. Data management is further complicated as UDI data is not static and must be updated based on changing regulatory requirements, such as market dates and registration status.
"Rimsys is the first automated UDI solution to fully integrate into a product’s registration lifecycle, ensuring that changing regulatory information is properly captured, registered, and maintained with the product and within government UDI databases like the GUDID (USA) and EUDAMED (EU)," said James Gianoutsos, Founder & President at Rimsys, "The current solutions landscape revolves around creating in-house, custom, and expensive projects with custom-built software or existing ERP software that were not designed to keep up with the fast-paced and ever-changing regulatory landscape."
In order to stay compliant and competitive, medical technology companies need to adapt quickly and flawlessly to the regulatory environment. As UDI requirements are rolled out across the globe, Rimsys’ UDI module seamlessly integrates with product registration data that is already actively managed within Rimsys, resulting in less maintenance, reduced costs, better compliance, and ultimately decreased time-to-market.
"We have already seen how our system has helped world-leading medical technology companies, such as Johnson & Johnson, Omron, and Terumo, digitize, automate, and navigate the global regulatory landscape,” said Brad Ryba, Chief Technology Officer at Rimsys, "As these requirements and demands continue to evolve, we are committed to ensuring that regulatory professionals can leverage the data integrations within Rimsys to more proactively manage their regulatory information."
Interested customers can schedule a preview of the new UDI module.

Rimsys named 2020 top technology company finalist by Tech 50 awards
Rimsys has been named a Finalist in Tech 50’s 2020 awards program hosted by the Pittsburgh Technology Council. The awards program, which was held November 12, 2020 at a virtual gala event, announced the winners and finalists. Rimsys was nominated as Innovator of the Year in the MedTech category, along with Sentact, Philips, and TeleTracking Technologies. In addition to the Company’s nomination, Rimsys’ Founder and CEO, James Gianoutsos, was nominated for CEO of the Year.
The awards program honors technology innovation in the region. Rimsys Inc., provider of the only holistic Regulatory Information Management (RIM) software for medtech is headquartered in Pittsburgh, Pennsylvania and was proud to be among the 2020 nominees. Rimsys was named a finalist this year, as Sentact, a provider of healthcare technology solutions for the patient care journey, walked away as the winner. Barbara VanKirk, CEO of IQ Inc., won this year’s CEO of the Year award.
"Rimsys is honored to be a finalist in both categories. Thank you to the Pittsburgh Technology Council for highlighting all of these innovative companies, and congratulations to this year’s winners," said Gianoutsos, "Rimsys is already used and trusted by leading companies, including Johnson & Johnson and Terumo, and we look forward to the near future where Rimsys’ automation of the global regulatory landscape is commonplace for medical technology companies and regulatory affairs professionals."
James Gianoutsos founded Rimsys in 2017, recognizing that while regulatory management platforms existed for pharmaceutical companies, medical technology companies face an entirely different set of challenges and requirements. Equipped with the expertise and drive to give regulatory affairs professionals a better way, he developed Rimsys: a RIM software built 100% for medtech companies.
Rimsys consolidates all the major functions of regulatory affairs on a 100% secure, cloud-based software, making product registration, standards management, essential principles/GSPR, and regulatory intelligence easy. Its newest integrated module manages global UDI requirements.
"It’s been an unparalleled year with unthinkable challenges," said Audrey Russo, President & CEO of the Pittsburgh Technology Council, "The COVID-19 Pandemic has tested everyone’s mettle in ways never imagined. This year’s Tech 50 is a testament to the fortitude, creativity, and innovation that exists across our industry, membership, and in every Tech 50 winner and finalist."
For a full list of finalists and winners, visit the Pittsburgh Technology Council’s website.
