


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.



The beginner's guide to the FDA De Novo classification process
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
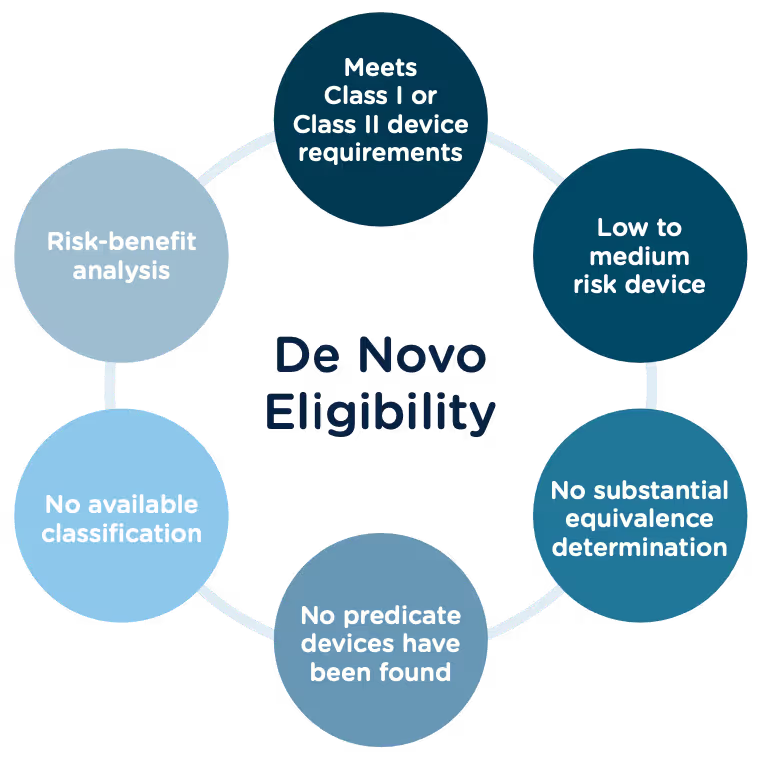
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
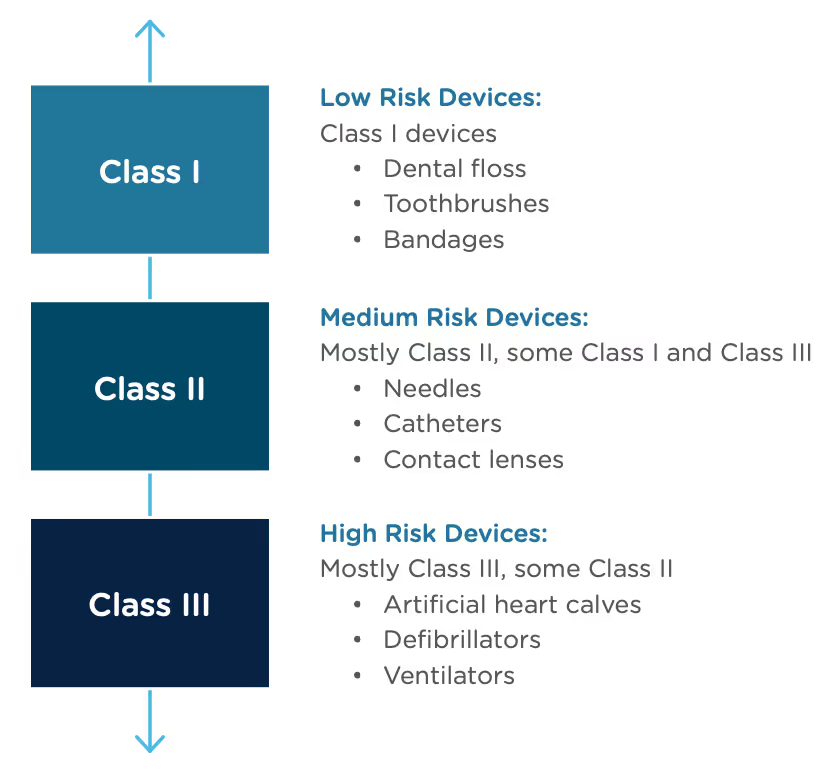
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.



%2520(1).avif)
Using EUDAMED as the Foundation for a Global UDI Strategy
If your company is selling medical devices in the European Union, you’re likely thinking about EUDAMED. If you’re not, it's time to start. Come January 2026, medical device manufacturers will be required to submit their UDI data to EUDAMED. EUDAMED’s data requirements and interconnectivity are complex. Not only is it easy to underestimate the time it will take to organize and verify UDI data, but many manufacturers are still using manual processes such as spreadsheets or internally built systems to manage this information. And with a number of organizations treating UDI data management and submission as an IT or supply chain process, some RA teams don’t even know where their UDI data is.
Getting your UDI data EUDAMED ready for submission by January 2026 is going to take some time, but there is a tremendous opportunity amidst the extensive preparation. With a little bit of foresight, RA teams can position the data output of their EUDAMED compliance project as the foundation for a global UDI program. Here are some reasons why using EUDAMED as the primary building block for a global UDI strategy will enable long-term success:
The product data needed for EUDAMED can be applied to current and future global UDI requirements
With many required data attributes and Basic UDI family groupings, establishing EUDAMED compliance for UDI is going to take some careful planning, detailed organization of current data, and careful consideration of your regulatory strategy for the European market. Many manufacturers need to start with the basics: locating current UDI data to understand how it is currently managed and transmitted to the respective regulatory bodies. This process is also going to require an assessment of your UDI data. Do you have information populated for all of the required attributes?Is this information current and accurate? Finding answers to these questions is going to take thoughtful collaboration among all stakeholders, but it’s also going to set your organization up with a solid data foundation that will help you meet additional market requirements.
It’s going to require an audit of your people, processes, and systems
Industry is focused on establishing UDI data in EUDAMED to align with current requirements for timing but transmitting your UDI interaction with EUDAMED is not a one-time process as it may be currently treated for other regulators. New devices, device changes, UDI data updates, and commercial availability may require EUDAMED updates or establishing new UDI profiles altogether. Are your current processes able to support you long-term? Manual processes are going to make it nearly impossible to keep pace and compliance, and internally built systems are going to require regular validation and subject matter expertise to keep up with evolving requirements and EUDAMED version updates. Taking the time to make sure you have the proper expertise and systems to maintain EUDAMED compliance will more easily allow you to scale your UDI management as your business grows and the regulatory landscape evolves.
The risks of non-compliance are too steep to ignore
Those who fail to submit UDI data to EUDAMED in line with the required timelines are at risk of audit findings, financial penalties, product delays, and worst of all, having to remove products from the market. Not only does product removal have lasting productivity, competitive, and economic impacts on the manufacturer, but it also decreases the availability of life-changing medical devices to end users. These risks are simply too significant to ignore and necessitate a comprehensive global compliance strategy that can meet the needs for EUDAMED and additional countries as global regulations change.
Additional regulators are establishing UDI requirements and big data technologies
As the value of UDI is recognized, More global markets are adopting UDI requirements to enhance the traceability of their devices and improve patient safety. For instance, Australia has established UDI requirements with mandatory compliance for data submission expected in the summer of 2025. Additionally, Rimsys expects the competent authorities from Switzerland and the United Kingdom to adopt their own UDI requirements following their exit from the European Union. Even though each regulator has varying requirements to some degree, organizing product data to meet EUDAMED’s robust set of data attributes and requirements will put you in a position to more easily meet additional global UDI requirements.
How Rimsys supports a global UDI strategy
Both the regulatory and technology requirements for UDI continue to grow in complexity. Rimsys has an integrated, automated UDI solution that provides medical device manufacturers the ability to manage their UDI data alongside their regulatory activities for total visibility and control.
Not only does Rimsys have the expertise necessary to help MedTech manufacturers navigate UDI data and transmission to EUDAMED for January 2026, but we're also dedicated to supporting MedTech companies with solutions and strategies that go beyond EUDAMED compliance to support a scalable and efficient global UDI program to meet the needs of evolving regulatory requirements. Rimsys provides MedTech manufacturers with a single-sourced UDI solution that allows them to manage their UDI data and status alongside regulatory activities.
Our novel Universal UDI® approach centrally stores common data attributes across global markets for simple management, reduced compliance risk, and increased efficiency. Using data established in Rimsys for EUDAMED as a baseline, we’re enabling MedTech companies to more easily meet additional global UDI requirements in a unified RIM solution that applies these common attributes between various market requirements (with more coming soon!):

Ready to increase your team’s efficiency and compliance with integrated and automated UDI management to support your long-term goals? Request a custom Rimsys UDI demo here!
%2520(1).avif)
Assessing RIM Maturity: Takeaways from our Expert Panel
Rimsys recently hosted a webinar, Assessing RIM Maturity for Your Regulatory Management Strategy, to help RA teams put together a practical RIM transformation plan for their MedTech company. Featuring industry leaders, Adrian Bishop of Boston Scientific, Brian Williams of KPMG, Steve Gens of Gens Associates, and James Gianoutsos of Rimsys, the discussion centered on the evolving MedTech regulatory information management (RIM) landscape, using the RIM Maturity Model Framework to guide your regulatory information management strategy, a RIM maturity case study, and AI’s role in regulatory information management. Here's a quick recap of the insights we discussed:
The Growing Need for MedTech RIM Modernization
The session underscored the increasing complexity of global regulatory requirements in the MedTech sector. From EU MDR to the adoption of EUDAMED, the shifting regulatory environment is pushing companies to modernize their RIM processes to ensure compliance and avoid operational inefficiencies. Steve Gens shared key insights from his 2024 World Class RIM Study research that by 2024, 52% of life sciences companies had fully adopted global RIM programs, a significant leap from 32% in 2022. However, many MedTech organizations remain in the early stages of RIM maturity, reflecting the need for process harmonization and robust digital solutions.
“The manual nature of spreadsheets and desktop authoring tools is no longer sustainable with all the different regulations and the global operating model that most companies have where you're trying to quickly get submissions out to different markets and then have to keep track of where your products are registered and where there are renewals, and where there are expiration dates. Having that information in informal spreadsheets and SharePoint sites is really not sustainable and creates a lot of compliance risks." - Brian Williams, KPMG
The RIM Maturity Model: A Framework for Transformation Progress
Our panel also explored the new RIM Maturity Model framework from Rimsys, which was built to help MedTech teams modernize their processes no matter where they are in their transformation journey.
“MedTech teams want to modernize their processes but don't know where to start from a business or data collection standpoint. They don't have a framework that takes their current processes into account." - James Gianoutsos, Rimsys
The RIM Maturity Model categorizes organizations into six levels based on their current regulatory information management processes:
- Level 0 - Unaware
- Level 1 – Aware
- Level 2 – Reactive
- Level 3 – Proactive
- Level 4 – Well Managed
- Level 5 – Optimized
Most MedTech teams fall between Levels 0 and 2, relying on siloed data and manual processes and reactive regulatory information management. Progressing toward higher maturity levels involves:
- Data Harmonization: Establishing consistent definitions and processes across business units.
- Process Transformation: Moving beyond "firefighting" modes to proactive regulatory planning.
- Technology Adoption: Implementing a RIM system that centralizes data, automates tasks, and facilitates cross-functional alignment. This is a critical step to reaching fully optimized regulatory information management.
“If you're considering a RIM adoption or are already going through one, make sure you understand your own expectations. You can't get to a spacecraft without without getting into a horse and buggy, Data collection and harmonization is milestone-driven and progression-based. You're not going to get to an optimized state on Day 1. But as long as you keep continuing to make well-informed decisions with your data and aligning internally on key terms in that data digitization processes, you will… get there." - James Gianoutsos, Rimsys
"As you're progressing, you need to be really thoughtful about the cost benefit of any initiatives that you're undertaking, Make sure that you have clear objectives that can be measured and that you can report value against and really just make sure that you create recognition with your management teams and with your operational teams that this is going to be a phased journey. You're not going to get there overnight and set expectations appropriately, but try to make measurable steps along the way. And as you do that, you start to build confidence, you build awareness and you build trust that you're delivering on the kind of the objectives that you set out to deliver. It's really a journey.” - Brian Williams, KPMG
Case Study: RIM Maturity Lessons from Boston Scientific
Adrienne Bishop from Boston Scientific shared her company’s journey toward RIM maturity. After previous attempts to implement global systems, the team learned to prioritize readiness, secure buy-in from stakeholders, and leverage external expertise.
“The added benefit of a MedTech solution is also that with these external partners, we can understand what is industry standard and that can help us with our change management.” -Adrienne Bishop, Boston Scientific
Key lessons they learned include:
- Start with Data: Define data fields clearly and cross-check sources to ensure accuracy.
- Limit Customization: Avoid over-customizing solutions that can hinder transformation; instead, adopt proven industry-standard processes.
- Invest in Change Management: Equip teams with the skills and mindset to embrace transformation.
Boston Scientific’s phased approach—beginning with foundational product registration data and expanding capabilities over time—demonstrates the importance of incremental progress. Boston Scientific, for example, has made strides in adopting a RIM system by focusing on transformation rather than harmonization alone, emphasizing the importance of cross-department collaboration and clear data governance.
"The solution we picked works for other people, so we can make it work for us. We also realized that what we were doing before maybe wasn't wrong, but it was definitely inefficient. We now have different inputs and different regulations. And now we need to change to get there." - Adrienne Bishop, Boston Scientific
AI and Automation in RIM
The panel distinguished the key differences between automation and AI, noting that while automation focuses on rule-based processes, AI serves as a good starting point for handling complex, abstract tasks. While AI can reduce time spent on tasks, it’s important for RA teams to verify the source information and the outputs for accuracy.
“There's a lot of anxiety about really what AI will deliver. But I think a lot of people that have been working with it, they see it as the virtual assistant or assisting. So it's more augmentation because there's a lot of concern.” - Steve Gens, Gens and Associates
Both automation and AI are important to regulatory modernization. Specific use cases include:
- Automation: Streamlining data ingestion, workflows, and compliance monitoring.
- AI: Mining historical regulatory data, collecting regulatory research, and enhancing regulatory intelligence.
Despite the attention around AI, the panel emphasized that its effectiveness hinges on high-quality, harmonized data. Poor data foundations can render even the most advanced AI tools ineffective, highlighting the need for a phased, data-first approach to digital transformation.
“There are some benefits you can definitely achieve with AI. But first and foremost data is going to be the most challenging yet the most effective way to get to that holy grail that I think everybody wants out of a regulatory information management system.” - James Gianoutsos, Rimsys
Looking Ahead: The Future of RIM As the MedTech industry advances
Achieving higher levels of RIM maturity will require strategic investment in technology, data, and talent. Organizations must treat RIM modernization as a holistic transformation, integrating robust processes and aligning cross-functional teams. With automation poised to play an increasingly significant role, companies that prioritize data governance and process maturity today will be well-positioned to leverage advanced technologies like AI tomorrow.
RIM modernization is not just a compliance imperative but a competitive advantage. By adopting a structured maturity model, focusing on data quality, and embracing RIM solutions like Rimsys, MedTech organizations can streamline regulatory processes, reduce risk, and drive innovation. If your organization is considering RIM modernization, now is the time to assess where you stand and take the first steps toward a holistic transformation.
For more insights, listen to the webinar recording here!
.avif)
5 Reasons It’s Time to Stop Managing Your UDI Data in Spreadsheets
UDI data management is continuously getting more complex. Not only do global markets have varying UDI requirements, but new markets are expected to come online in the coming months. Additionally, the mandatory application of EUDAMED will begin in January of 2026, meaning that MedTech manufacturers selling products in the European Union will have to submit their UDI information to EUDAMED.
Submitting UDI information to EUDAMED will require careful and thorough preparation to ensure that manufacturers have all of the required data attributes necessary to meet the EU’s requirements and that they have a strategy in place to organize, review, and submit all of their information.
MedTech regulatory affairs processes are largely manual and siloed, and UDI management is no different. Many teams are still managing their UDI data in complex spreadsheets. While the forthcoming mandatory EUDAMED requirements have prompted many manufacturers to start planning more comprehensive UDI strategies, managing a high volume of UDI data in spreadsheets is unsustainable long term.
Efficient and scalable UDI management requires deep expertise, collaboration, and full visibility into your products’ global regulatory activities that spreadsheets simply can’t match. Here are 5 reasons it’s time to move away from managing your UDI data in spreadsheets:
1. They’re setting you up for compliance risks
UDI management involves a high volume of data across different markets and regulator IT systems. Not only are the assembly and maintenance of those data in a spreadsheet prone to human error, but they don’t enable the regulatory expertise necessary to ensure that your requirements for each applicable market are up to date. Further, siloed spreadsheets aren’t connected to systems that help you unlock regulatory insights. The responsibility is yours to make sure your spreadsheet has current, accurate, and reliable information.
With cumbersome version history and change tracking (or lack thereof), spreadsheets also lack the traceability needed for audits, making it difficult and time-consuming for MedTech companies to effectively demonstrate compliance. This not only adds time to the audit, but opens your organization up for audit findings, financial penalties, product delays, and revenue loss.
2. They’re time-consuming and difficult to manage
When multiplying the markets you’re selling in by the number of products you’re selling, it’s easy to see how complex UDI spreadsheets can get. Since there’s no automation to ensure the attributes and data in your spreadsheet are up to date, it’s up to you to validate it periodically and research the updated requirements. If you do establish a process to validate that UDI data, this adds critical time for Medtech teams that are already tasked to execute faster with limited team members. If you do not periodically validate UDI data, you take on an increased risk of non-compliance.
Additionally, new market regulatory and IT requirements make managing UDI information even more difficult each time a new market is added. With several countries expected to mandate or enact new UDI requirements and systems over the next couple of years, the complexity is only going to grow with time.
3. They limit collaboration across your team
Multiple stakeholders working within the same spreadsheet can easily create confusion. Not only is it difficult to track changes, but there’s a likelihood of overwriting critical information. This can not only lead to the compliance risks mentioned above, but spreadsheets also impact your team’s ability to effectively collaborate. Team members often duplicate the spreadsheet and work off of different versions when there are limited controls in place to prevent this from happening.
On a strategic level, spreadsheets impact your team’s ability to share knowledge with one another. There’s no way to effectively share detailed notes and tasks, and there’s also limited functionality to track transmission status and troubleshoot transmission errors with your team.
4. They don’t provide full visibility into your products’ selling statuses
UDI spreadsheets are usually designed for one purpose, UDI data management. However, UDI is just one facet of obtaining market clearance for your product. The product must also be approved and registered in each respective market. Many manufacturers have multiple teams and spreadsheets to track regulatory activities where UDI responsibilities may lie outside of the regulatory team. This approach limits their ability to get full visibility into the status of their products and integrate this information with other relevant applications across the business. With regulatory information scattered and siloed, it’s easy to miss the connection between UDI data and upcoming expirations and renewals.
5. They don’t easily scale with your business and growing market requirements
As you create new products and bring existing ones into new markets, do you have a plan in place to help you scale effectively? The regulatory activities along with the expanding UDI requirements associated with new product introductions and market expansions are high enough, and spreadsheets simply can’t keep pace with business growth.
Moving your UDI management off spreadsheets is going to set you up for a successful, long-term UDI program. MedTech companies not only need to be aware of the forthcoming mandatory EUDAMED application timelines, but they also need a sustainable solution as additional UDI requirements come online. Finding a solution that’s built with the complexities of UDI data management in mind, automates time-consuming, manual processes, and offers complete visibility into all of your regulatory activities is going to help you ensure that your UDI program is compliant and scalable to meet your evolving business needs.
The Rimsys RIM platform allows you to manage your UDI program in a single-sourced, unified, and connected solution that enables you to track your UDI activities alongside your product registrations and selling status to give you confidence in your compliance. Additionally, Rimsys has a novel Universal UDI approach that captures common attributes across various market requirements to reduce administrative burden and help you submit your UDI information consistently and with ease.
Want to see how a purpose-built MedTech RIM solution can offer you a compliant, scalable, and comprehensive approach to UDI management? Request a demo to see Rimsys in action!
.avif)
Rimsys secures $5 million in growth financing round
Rimsys recently secured $5 million in a growth financing round from Global Opportunity Pennsylvania Fund II, L.P. (GO PA Fund), Riverfront Ventures, and existing investor, Innovation Works. GO PA Fund, Riverfront Ventures, and Innovation Works have a strong commitment to fostering innovation and creativity in Pennsylvania, which makes them great financial partners for Rimsys.
“We’re ecstatic to have support from GO PA Fund, Riverfront, and existing investors as we enter a new growth phase at Rimsys,” said James Gianoutsos, Rimsys Founder and CEO. “Our next phase of growth builds on our mission to increase accessibility to life-changing medical technologies with a focus on regulatory intelligence, submissions management, and UDI enhancements and thoughtfully incorporating advanced technologies like AI into our roadmap to meet growing demand and changing global regulatory requirements. We are grateful to work with firms that are committed to nurturing innovation in the Pittsburgh region, which has given so much opportunity to us.”
Our growth financing will help ensure that Rimsys provides a world-class, unified regulatory information management (RIM) solution for the medtech industry and that it meets the challenges that come with a frequently evolving regulatory landscape.
Those interested in learning more can read the press release from GO PA Fund and a recent Post-Gazette interview with our founder, James Gianoutsos:
ABOUT GO PA FUND
Formed by Ben Franklin Technology Partners of Southeastern Pennsylvania, with collaboration from Ben Franklin of Northeastern Pennsylvania, Ben Franklin of Central and Northern Pennsylvania, and Innovations Works from Southwestern Pennsylvania, the GO PA Fund invests in technology-based ventures throughout the Commonwealth of Pennsylvania. The GO PA Fund primarily invests as a follow-on fund to companies selected from Ben Franklin’s statewide portfolio of more than 600 burgeoning ventures throughout Pennsylvania. The Fund leverages Ben Franklin’s best-in-class multi-year/multi-round due diligence to ensure access to qualified and vetted opportunities while minimizing costs of investment. Visit us at gopafund.com.
ABOUT BEN FRANKLIN TECHNOLOGY PARTNERS OF SOUTHEASTERN PENNSYLVANIA
Ben Franklin Technology Partners of Southeastern Pennsylvania (“Ben Franklin”) is the Philadelphia region’s Partners with a Purpose. Nationally ranked among the most active seed and early-stage investors, Ben Franklin helps high-growth innovative enterprises plant and nurture their roots, creating both immediate connections and lasting economic growth. The nonprofit has supported more than 2,000 companies to deliver an impact of more than $5 billion and 32,000 jobs in the Philadelphia region. Whether in tech, life sciences, manufacturing, or industries and breakthroughs yet discovered, Ben Franklin works to raise the community of innovation higher, to benefit present and future generations of Pennsylvanians. Visit us at partnerswithapurpose.org, or follow us at @bftp_sep.
ABOUT INNOVATION WORKS
Innovation Works is one of the most active early-stage investors in the country and the most active in Pennsylvania. Since its inception of the seed fund in 1999, Innovation Works has invested in over 760 companies that have gone on to raise $3.3 billion in follow-on funding. Portfolio companies have generated and retained over 20,000 jobs in Pennsylvania. Innovation Works is part of the Ben Franklin Technology Partners network, which has catalyzed economic growth over the last 30 years by providing access to capital and networks that help foster innovation and technology-based economic development in Pennsylvania. Learn more at innovationworks.org.
ABOUT RIVERFRONT VENTURES
Riverfront Ventures is an early-stage venture capital fund based in Pittsburgh, PA focused on investing in early-growth stage tech companies. Learn more at riverfrontventures.com.
ABOUT RIMSYS
Rimsys is improving global health by accelerating delivery and increasing availability of life-changing medical technologies. Rimsys Regulatory Information Management (RIM) software digitizes and automates regulatory activities, helping medtech regulatory affairs teams to plan more effectively, execute more quickly, and confidently ensure global regulatory compliance. Rimsys is designed around medtech workflows and supports a full breadth of regulatory activities including registrations, submissions, UDI, essential principles, and standards management in a single, integrated platform. For more information, visit www.rimsys.io.
_thumbnail.avif)
Insights from the Gens and Associates Executive Podcast with James Gianoutsos
Rimsys Founder & CEO, James Gianoutsos, was recently a guest on the Gens and Associates Podcast, a series dedicated to regulatory trends, topics, and insights from industry thought leaders. During the interview, James and Gens and Associates Managing Partner, Steve Gens, shared their unique founding stories and discussed digital transformation in the medtech industry.
One particular aspect James and Steve discussed is the role of AI in medtech regulatory affairs. Historically, the medtech industry has lagged behind pharma in digital adoption by 10-15 years. As the medtech industry takes on digital transformation initiatives, some medtech companies pursue AI solutions first. Through his extensive experience helping medtech companies embrace and adopt digital change, James states the importance of starting with efficient regulatory information management. Listen to the full interview to learn why setting a proper data foundation is critical to leveraging AI successfully along with:
- How AI will evolve in medtech regulatory affairs
- The importance of data governance in medtech digital transformation
- Why partnering with a medtech-focused RIM provider like Rimsys is essential for a successful transformation
- What's next on the Rimsys product roadmap as the company enters its next phase of growth, including enhancements to Rimsys Intel
Find the full interview on the Gens and Associates website, Spotify and Apple.
We'd like to thank Gens and Associates for featuring James and for the opportunity for him to share his thoughts about medtech digital transformation with the Gens and Associates community!
About Gens and Associates
Gens and Associates is a boutique Life Science management and organizational consultancy specializing in strategic planning and roadmap development, industry benchmarking, regulatory information management, organizational transition management, and working with leadership and project teams to accelerate change and value realization. Learn more on the Gens and Associates website.
TRANSCRIPT:
Steve Gens: Welcome to the gens, an associates executive podcast series where I have one-on-one conversations with leading executives that represent the Regulatory software and services sector, to learn more about how their organizations are supporting, and more importantly innovating this space. So, Steve gens here, managing partner of Gens and Associates today, I'm happy to be speaking with James Gianoutsos, founder and CEO of Rimsys. So welcome, James, I've been looking forward to this conversation and, you know, as we have this executive series, it's kind of rare to have a founder, you know, founding Gens and Associates in 2005, seemed like a very risky proposition and it's different when you're founding something versus just being a CEO of existing organization. So, before we get started with Rimsys being a fairly new company, I think you were founded in 2017. Could you give our listers a brief history of Rimsys and yourself and how you support the me tech sector?
James Gianoutsos: Yeah, absolutely. And before I start off, just thanks for having myself on board and being a presenter here. And yeah, for everybody who does not understand the med tech arena or know Rimsys, my name is James Gianoutsos on founder and CEO of Rimsys. And I've spent the last 17 plus years in the Regulatory and quality industry.
And so I started my career at Philips and worked for several small and medium sized medtech manufacturers. I've had, the experience of actually managing global Regulatory operations, doing global submissions, moving 600,000,000 dollar manufacturing locations under consent decrees in the midst of the EU MDR and I VDR transitions. And so I've also had a really nice breadth, of medtech background as well, medtech product background, I should say all the way from c- pap devices to internal surgical adhesives even on the consumer side such as pacifiers, incubators, you name it.
I've really had a really diverse in broad experience level with medtech itself. And so back in 2017, I was actually working for a small medtech manufacturer. And fortunately, unfortunately, I was laid off of that position. It was something that I know that there's a lot of medtech manufacturers that are kind of doing the same thing in this year last year, I should say. And… and it was, you know, that would happen on May 31, 2017, at noon and I started Rimsys at 1 PM that same day. It was just something that I've always had in the back of my mind's something that I always wanted to pursue.
And really looking at the landscape back then there were only pharmaceutical RIM providers on the market. And so there was nothing really catered to, the medtech industry. Medtech and pharma aren’t uniform and aren’t just worlds but universes apart in terms of regulatory complexity, how you get products to market, how you maintain those products on the market and just the regulatory pathways and workflows around that. And so really started my endeavor hiring a couple developers offshore.
And, and to your point, you know, it's definitely different to be a founder than just a CEO because you're so heavily invested and so heavily involved that's you know, over the last five to six years… seven years is to say it's been a really cool experience to be personally involved with this development and working with the largest tech manufacturers in the world to help develop our solution and improve the regulatory workflows. So at the end of the day, they can get the products to market faster and keep those products on the market.
Steve Gens: Excellent. So thank you so much for that good introduction. And then again from one founder to another, I still remember I had a fold up desk in a spare room with a laptop, my cell phone, you know, one customer, one contract and, you know, we're starting our twentieth year and have over 100 global customers. Like, I could never imagine back then where it would be but, you start someplace, you have a vision, you see a need, you go after it. You, you just get the best team around you and you just do it. So, so congratulations, you had a very successful lift off, and that's where I'd like to start.
I know when we first got introduced and started covering you about four years ago. And, I think it's on your website too is, you know, that context because again, we support both the biopharmaceutical and tech and they are very different although there's growing combinational products, right? So they're starting to be a little worrying, but there they are two different worlds or universes. Kind of in your case, you know, I know you say, yeah, we build this by Regulatory affairs, you know, professionals, there's a better way to do this. And also like the other terminology that a lot of people use big words like transformation and all that. But in Regulatory, it's a major monetization and simplification. And I know on the medtech outside, organizationally you know, they tend to be a lot more distributed design center. So in some ways you have a different set of complexity. So we've been tracking you for about four years now, we've watched your growth. We get those regular annual updates, but I think one thing our customers would really be interested in is, you know, why do your customers pick Rimsys? You know, what differentiates you from, the competition?
James Gianoutsos: Yeah, there's a lot to unpack from that question. And I'm, happy to dive in a little bit more to it. You know, one of the primary things that we do and understand is the medtech industry period. And so from day one, you know, this is why our company exists to support medtech. There's you know, the whole thesis around Rimsys around our solution and our product, in the industry historically medtech is 10 to 15 years behind pharma from a digital transformation standpoint.
Medtech has been undergoing this digital transit initiative over the last several years and since then, we've really taken off because especially at the enterprise level standpoint, and the complexity around the workflows, the understanding of how the, in relation between the data elements of not just, you know, regulatory information, but of how the products are associated to those registrations to certificates to really all the entire Regulatory product life cycle really differentiates us because, we understand, those nuances better than anybody in the industry period.
You know, we've a we've had a lot of success because it feels like, the pharma industry is kind of waking up to it. We've actually seen some pharma companies try to come into medtech and actually failed miserably because, you know, I would say with 100 percent conviction that nobody wants a pharma RIM with the medtech label slapped onto it. And what allows us to be successful in that is that we partner deeply with our enterprise customers to understand those nuances, and we're adaptable to those changing regulatory needs. We are a relatively young company. But we are broadly and vastly experienced in that med tech space. And so that's really been one of our competitive advantages is just understanding the space like nobody else.
And you mentioned combination products and what's really unique about medtech is that, you know, combination products at the end of the day are a drug coded stint or some type of prefilled syringe, those are a medical device. And based on the primary mode of action, some other items that are typically more medtech forward devices. And so that creates an interesting opportunity for Rimsys because, you know, drugs are drugs at the end of the day, it might have different dosages, but the varying aspects of where that do can be put into the system or put into your system. I should say… your body is vast and a lot of those have or need a medical device to deliver that.
And so, the variations and complexity around that makes for Rimsys to be at the forefront and leader of that combination product standpoint. And a lot of our customers have actually adopted Rimsys even more heavily because of that, not just on the medtech side but on combination side.
Another interesting item that we're seeing is that we are also because medtech as it is so vast and so complicated and complex from the workflows. You know, we're product centric and that has the adjacent categories as well where you can go into veterinary biocides, biologics consumer side combination because at the end of the day, the framework in the structure is generally the same. However, the regulatory pathways might be different. And so, that bodes well with our client base because a medical device manufacturers product portfolio, is ever-changing but also becoming more broad in nature as it expands itself into its new markets.
Steve Gens: Yeah. Thanks for that overview. And I've been taking some notes on that. And I've seen kind of on the biopharmaceutical side, some of the RIM providers, hey, we should do medtech and, you know how I maybe I'm looking at this naive is like in the biopharmaceutical generics, you know, it's more of a data paradigm where, your life is an engineering paradigm and as opposed to like one to one with the health ministry or the health authority, you could have one to many in each country, right?
James Gianoutsos: Precisely.
Steve Gens: In your submission, so it's that extra kind of permutation if you will of complexity and maybe another podcast, maybe we'll do later on too. I was just thinking about, you brought up the stint in earlier in my career. I think, you know, I was a Johnson and Johnson guy and one day, you know, one of the pharma scientists was, you know, talking with one of the device scientists and they came up with a stint that must be like 20 years ago. But maybe another conversation, just the emergence of software as a device. You know, that's a big interesting topic. But that's venture on a little bit more so.
And I know we talked about this kind of, the other day, you know, about monetization of regulatory systems and processes. And there's such an intense focus. The big word this day is AI, right? But there's AI and automation, and sometimes people confuse, the two. What's Rimsys doing as far as helping your customers, and maybe just in the very near term. So maybe tactical things we're you're doing either with AI or automation or the combination. And what does the longer term look like?
James Gianoutsos: Yeah, it's a great question. And to your point, I think there's a couple items that, I specifically want to address there because at the end of the day, you know, AI, is this thing that's being used from a marketing standpoint or just from a general industry standpoint in general, right? And it's this idea that these systems or software solutions can help do the work for you. Not, you know, it's not gonna take anybody's job, but there are ways that it can definitely help make you more efficient.
You know, what's particularly interesting in the medtech digital digitalization transformation initiative that's going on right now is that, you know, medtech in itself is 10 to 15 years behind the times period. And so especially with these large enterprise level companies. And so, when you're in a horse and buggy, which is what the med tech industry is today. And we're trying to get into a spacecraft, of course, I think, the logical and the initial item in thinking is that, okay, we can just go straight to AI, but really at the end of the day AI is only as good as the data that is trained on.
And… phase one of the company has always been the information management period of medtech in the organization. In getting that, right, we've invested so much time and money and collaboration with our partners of how product attributes, UDI attributes, regulatory… attributes. And how all of that plays well and interlays with one another because if you don't get that fundamental framework, you're gonna have a hard time. You might do some really interesting things with AI, but you're gonna have a hard time keeping that information organized in the manner that you will need to have it organized to do things later on.
And so, what's really been interesting is that we're about complete with our phase one of the company, meaning that the information and organization and complex workflows have a majority of that has been addressed, meaning that we have now reached this pinnacle where we're gonna go into phase two of Rimsys.
Pase two has really been in and is all about the Intelligence. And so if you think about everything we've done in the phase one of the company where it's the information management, the we're call it the data layer. Now, we're entering the phase two, we're now we can overlay intelligence and market information directly over that data layer, so you can do some really fun and interesting and innovative things with that data. And so from an AI standpoint, we actually have some things coming down the pipeline at the end of this year, early next year, that's really going to help one with data ingestion in our system.
You know, we have companies that have 300,000 products and 249 countries globally and that's just one company, right? And so there are some really interesting things we can do with data ingestion and data maintenance from our standpoint. But also there's some really interesting things we're gonna do with market intelligence notifications of, you know, regulatory changes in the market as well as some things that are coming through with submissions as well. And, and we have a really nice road map played out that in the next six to 12 months that it'll be, it's gonna be really cool to see some of those things come to fruition here.
Steve Gens: Yeah. There's there's a lot. I just took a lot of notes on that on this and just a few comments I think, you know, for our listeners too, just another layer of complexity between, the med tech and pharmaceutical is, besides the things you've mentioned, I've mentioned, you know, you're dealing with a class one, two or three, you know, device. So like the one client with, you know, all those products. It's kind of reminds me of the consumer side and JNJ, it's like you have Tylenol as a product but you have so many variations then different names of it in so many different countries, mind explodes just trying to manage the label.
You know on that, I think the other thing on the probably where you guys are more ahead on the medtech side is there's this nirvana of embedded reg intel, you know, where, you know, the reg intel actually directs the workflow you know, as opposed to, the user based on whatever regulatory activity you're working on. If you're doing a renewal in Thailand, for example, it has the reg intel and it knows what to do so.
The same thing too, some people are scared of AI is going to replace, you know, my job, but I think you know, more and more people realize it's a virtual assistant or a writing assistant where the AI might do the first version, one of a document, you know, the boring stuff. And then the expert medical writer would actually take it, you know, with their scientific knowledge.
And the last thing I wanted to comment to because we just finished up our very large study, you know, with AI, it's only as good as the data we've proved out. And I don't know how much of an issue. It is just like having the highest level of data quality and regulatory, but, you know, a lot of folks on the biopharmaceutical side, it's like well, you know, should we enter it centrally, decentralize, hybrid at that time? How the data is entered into data quality where that is not linked. We've proved it out that having those really good data quality practices, the data governance, have that in place and that's a direct correlation. We call those the data assets, you know. And then there's a shiny activity. If you have the right skills and the right KPI, you put those four together. And that's where the magic happens, right?
So, the last question, and actually from founder to founder, you had the sparkle in your eye when you thought about this and you actually pulled the trigger. So instead of, you know, kind of having the CEO voice, but from the founder's voice, what excites you most about Rimsys in the coming years. So, where is the company going?
James Gianoutsos: It's such a loaded question because I feel like there's so many amazing things to do. I mean, we're still at the beginning of this whole thing. And my mind has been going since, you know, 10 years ago when I first arrived this, you know, to 2017 when I first started putting… pen to paper.
And, you know, having all these tools and things that I wish I had when I was in industry would have been absolutely amazing not just from a jobs perspective standpoint, but from the company perspective standpoint because the things that regulatory does has a direct impact on the revenue as well as getting those products to market for the patients need the most and maintain those products on the market for the patients that need in the most. And so, there's a high degree of vested interest to get those products and keep those products on the market as a regulatory professional, honestly just as a human being.
And so, the things that I'm excited for are the new Rimsys Intel that we're gonna be continuing to advance here in the next six to nine months. And there's gonna be some, really exciting things, that I think there's some new adjacent product regulatory life cycle items that we can get into. So, there's always the premarket, on-market, and post-market. And we really haven't even touched post-market yet. We've really been concentrating on the on-market and premarket aspects of things and especially getting UDI right. Because at the end of the day, those udi attributes are needed for a lot of the post-market activities that are on gonna be a regulatory requirement or already a regulatory requirement.
But then two, from a reporting standpoint, you're gonna need to maintain those products in the market. And so, there's this continuum of information management that we're gonna be continuing to do and gather, and address, and then, the layering on top of that Rimsys, Intel is absolutely gonna be a game changer because nobody really does RIM like Rimsys, and, you know, we've had the luxury of building this from the ground up specifically dedicated to the medtech industry. And that has so many more advantages than trying to, reposition or retransform an existing system to medtech because it just, it doesn't translate. And so, I'm really excited, for some of those aspects.
Steve Gens: Yes, indeed. You know, a very exciting, and also just an amazing journey in just seven years. So, and it seems like it's a very bright future, you know, for you. So, so thanks again for your time, you know, some very Rich and insightful discussions, you know, and it's great with our listeners and some of them are many of them are biopharmaceutical, you know, learning a little bit more about the med tech side.
But in the lens and why I thought this was so important and we kind of touched on it just the growing a portfolio of combination products - It's just really merging our latest data. Had 60 percent of the companies with their product portfolios have combination products. So that's something that's growing. So certainly listeners, if you're on the biopharmaceutical side, you know, maybe you have combo products or a device division, you know, definitely give them a look. Yeah. So as we kind of, you know, close up, some of our listeners might want to get a hold of you. So what's, the best way to contact you? I don't know if it's through the website or LinkedIn or, what would you suggest?
James Gianoutsos: Yeah, I would say definitely get our website: www.rimsys.io. You can schedule a demo and you can actually just put a Linkedin request to me directly. I love talking shop. I love talking specifically around regulatory complexities and some of the issues that you're experiencing firsthand because at the end of the day, those help continue to expand our system capabilities and serve, the medtech market. You know, one of the things that I'm really proud of with Rimsys is, you know, we already have 40 percent of the top 10 medical device manufacturers globally and we're expanding more. And this is a really exciting time for the industry as well, as well as, for Rimsys as we enter into this new phase of growth.
Steve Gens: Excellent. And, you know, indeed. And I think, you shared, you know, the other day, well name of another very impressive logo thatat you're gonna be starting to work with there. So before I say goodbye to the listeners, you know, we're both fouders and have that in common. But I would be remiss here. Maybe this is more for us listeners that you're based in Pittsburgh, Pennsylvania. I grew up in central Pennsylvania. So it's the black and gold. I know we talk about the Pittsburgh steelers, the, you know, NFL football seasons about ready to kick off. So hopefully the black and gold, the Pittsburgh steelers are gonna do well. I know we another thing that we have in common. Yeah.
So with that said with our listeners, if there's any questions you have for the Gens team use our contact page off of our website or similarly just reach out, I'm on Linkedin quite a bit and please enjoy our other podcasts. We actually just reorganized our whole podcast web page to have a section for this executive series, our world class RIM research, and then a third as we have different subject matter experts, really talking in detail about some of the key issues that industry works on today. So again, James, thanks a lot for your time and maybe in another six or nine months, we'll have you come back and see where yourself and Rimsys are at.
James Gianoutsos: Sounds great. Thank you.
.avif)
Rimsys NPI is here: Streamlined new product introduction for faster market entry
Rimsys is excited to announce a new feature to help medtech regulatory affairs professionals gain faster market entry: New Product Introduction (NPI). The Rimsys NPI solution significantly accelerates decision-making and reduces time to market by centralizing decision-making and automating time-consuming, manual processes. NPI also expands Rimsys’ extensive list of regulatory workflows that help medtech regulatory teams manage their products across the regulatory lifecycle.
New product introductions typically involve one of two important decisions: deciding which market to take a new product or, more commonly, deciding where to take an existing product to market next. Both involve careful planning and collaboration across numerous internal and external stakeholders. Regulatory teams need to assess the amount of time, resources, and costs needed to enter each new market. This process often involves careful examination of the product’s existing registrations. There are also business considerations they need to make including forecasting expected revenue gains for each market.
Traditional approaches to new product introductions are time-consuming and manual. There's no centralized place to manage regulatory information, making it difficult to view the product’s existing registrations, collaborate efficiently, and forecast effectively. Additionally, these processes are often complex, involving numerous spreadsheets and disjointed systems that drastically increase operating costs and slow decision-making – and ultimately time to market.
The Rimsys NPI solution addresses the common challenges companies face when doing a new product introduction project. By providing complete visibility into all regulatory information in a single platform, companies can streamline their processes and improve efficiency with:
- Centralized data management – Streamline the request and approval workflows needed to place a product in a specific market in one platform
- Enhanced collaboration - Collaborate with all relevant internal and external stakeholders directly within Rimsys
- Automated workflows - Automatically create registrations directly from the project
- Forecasting and decision support - Forecast expected revenue from NPI projects directly in Rimsys, and realize revenue gains faster
- Easier market entry - Reduce manual data collection efforts. Stay updated on important timelines, delegate tasks, and keep track of progress in a streamlined project
With its new NPI solution, Rimsys is excited to continue its mission of increasing the accessibility of life-changing products by giving medtech RA teams a centralized collaboration hub for NPI projects backed by the automation necessary to more accurately forecast, speed decision-making, and remove market entry barriers.
Ready to revolutionize your company’s NPI process? Request an NPI demo at rimsys.io/demo.
