


Featured
Rimsys Announces Rimsys AI to Eliminate Repetitive Tasks and Enhance Decision-Making for MedTech Regulatory Teams
Rimsys, the leading Regulatory Information Management (RIM) platform for the MedTech industry, today announced the launch of Rimsys AI, a suite of embedded artificial intelligence (AI) agents.


An overview of 21 CFR Part 820 - quality systems for medical device manufacturers
What is 21 CFR Part 820?
21 CFR 820 is the FDA federal regulation that pertains to quality systems for medical device manufacturers, and it is part of the agency’s set of Current Good Manufacturing Practices (CGMP) for industry. Also referred to as the FDA’s quality system regulation (QSR), the regulation defines design controls and quality processes at all stages of device development in order to ensure that all medical devices marketed in the United States are safe and effective.
21 CFR 820 consists of 15 subparts, which define quality system requirements for each stage and function within the medical device manufacturing process. We define each subpart below.
Federal regulations are organized as Title → Chapter → Subchapter → Part, which means that 21 CFR 820 is short-hand for:

21 CFR 820 vs ISO 13485
ISO 13485 is the de facto international quality system standard for medical device manufacturers, but this is not currently the standard in the United States. While Part 820 and ISO 13485 are structured differently, they have no conflicting requirements. Therefore, companies that are marketing medical devices in the U.S. and in other markets will need to comply with both ISO 13485 and the FDA’s QSR, as defined in 21 CFR 820.
However, the FDA is moving towards harmonizing these standards, and on February 23, 2022 issued a proposed rule to amend the QSR to align more closely with the international consensus standard for Quality Management Systems, primarily by incorporating reference to the ISO 13485 standard. The FDA has published FAQ’s about the proposed rule.
21 CFR Part 820 Requirements
Part 820: General Controls (subpart A)
The General Controls subpart contains three sections providing general information about the regulation, including the scope and applicability along with key definitions.
Scope
The regulation defines current good manufacturing practice (CGMP) requirements governing the methods, facilities, and controls used for the “design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices intended for human use." Specifically, this subpart defines:
- Applicability:
The requirements of this regulation are intended to ensure the safety and efficacy of all finished medical devices intended for human use that are manufactured in or imported into the United States. Manufacturers that are involved in some, but not all, manufacturing operations should comply with those requirements that are applicable to the functions they are performing.
Exceptions:
- This regulation does not apply to manufacturers of medical device components, but such manufacturers are encouraged to use this regulation as guidance.
- Class I medical devices are exempt from the Design Controls defined in this regulation, except for those listed in § 820.30(a)(2).
- Manufacturers of blood and blood components are not subject to this regulation but are subject to Biologics good manufacturing practices as defined in Subchapter F, Part 606 of the regulation.
Definitions
This section of the regulation contains definitions for a number of terms used throughout the document. The following are the major definitions related to quality records:
- Design history file (DHF): A compilation of records that describes the design history of a finished device.
- Design input: The physical and performance requirements of a device that are used as a basis for device design.
- Design output: The results of a design effort at each design phase and at the end of the total design effort. The finished design output is the basis for the device master record. The total finished design output consists of the device, its packaging and labeling, and the device master record.
- Device history record (DHR): A compilation of records containing the production history of a finished device.
- Device master record (DMR): A compilation of records containing the procedures and specifications for a finished device.
Quality System
The section of the regulation sets the basic requirement for a quality system by stating that “Each manufacturer shall establish and maintain a quality system that is appropriate for the specific medical device(s) designed or manufactured, and that meets the requirements of this part.”
The term “appropriate” is used throughout this regulation and can be open to interpretation. A manufacturer, however, should assume that all requirements are appropriate and applicable except in cases where non-implementation of the requirement can be shown to have no effect on the product's specified requirements or ability to carry out necessary corrective actions.
Quality system requirements (subpart B)
This section of the regulation defines the overall responsibilities and the resources required for the management of the quality system.
Management responsibilities
Executive management is responsible for establishing a quality policy and ensuring adequate resources to effectively maintain and manage the quality system. In addition, management is responsible for establishing a specific quality plan, consisting of relevant practices, resources, activities, and procedures.
Quality audit
Periodic audits of the quality system are required to be conducted by personnel not directly responsible for the activities being audited. The dates and results of each audit need to be documented, along with the results of the audit. It is expected that corrective actions and, when necessary, reaudits, be performed for any identified noncompliances.
Personnel
Manufacturers are responsible for assigning sufficient personnel with appropriate experience and training to perform all tasks required by the quality system plan.
Design controls (subpart C)
Manufacturers of all class II and class III medical devices, along with the specific class I devices listed in paragraph (a)(2) of this regulation, are required to establish design control procedures that ensure design requirements are met as specified.
Design controls shall define:
- Design and development planning - Plans that describe the design and development activities, and responsibilities for these activities and their implementation.
- Design input - Procedures that ensure design requirements are appropriate and address the intended use of the device.
- Design output - Procedures that document design output, including acceptance criteria, so that conformance to design input requirements can be adequately evaluated.
- Design review - Formal and documented reviews of the ensign results that include participation from representatives of all.
- Design verification - Procedures for verifying the device design that confirm that the design output meets the design input requirements.
- Design validation - Procedures for validating the device design, ensuring that devices conform to defined user needs and intended uses, and including testing of production units under actual or simulated conditions.
- Design transfer - Procedures to ensure that the device design is correctly translated into production specification.
- Design changes - Procedures for identifying, documenting, validating, and managing the verification and approval process of all design changes before they are implemented.
- Design history file - A design history file (DHF) is required for each type of device and should include or reference the records necessary to demonstrate that the design was developed in accordance with the approved design plan and device requirements.
Document controls (subpart D)
Medical device manufacturers are required to put in place document controls for all documents required in this regulation.
Document approval and distribution
One or more people must be assigned to review and approve documents prior to issuance. The approval must be documented, include a date and the signature of the approver, and be made available at all locations where applicable. Procedures must also be in place to ensure that obsolete documents are removed and/or prevented from being used.
Document changes
Similar to document approval procedures, changes to documents must be approved, reviewed, and documented. Records of all changes must be maintained.
Purchasing controls (subpart E)
To continue reading this Regulatory Brief, including a definition of the remaining subparts and a comparison of 21 CFR 820 to ISO 13485, please download the full brief.



CE marking guide for medical devices in the EU
This article is an excerpt from the CE marking guide for medical devices in the European Union.
Table of Contents
- What is CE marking?
- Why is CE marking important?
- CE marking responsibilities
- What countries require or accept CE marking?
- Which medical devices require a CE mark?
- Technical documentation
- What are the costs associated with CE marking?
- How do you apply the CE marking?
- CE mark and UDI
- Does the CE mark expire?
- Do I need to CE mark my software?
- Final steps
CE marking is a symbol that consists of “CE, “ which is the abbreviation of the French phrase "Conformité Européene" meaning "European Conformity". The term initially used to describe “CE” was "EC Mark" but it has officially been replaced by "CE marking" according to the EU Directive 93/68/EEC. CE marking is used in all EU official documents, although you will still see "EC Mark" being used in common language. If you are using EC Mark in your documentation, you should change that terminology to CE marking in the future.
The letters ‘CE’ appear on many products traded on the Single Market in all the member states of the European Union plus Iceland, Liechtenstein, Norway and Switzerland. Simply put, The CE mark is a mandatory compliance mark, informing the consumer that the product is compliant with all applicable EU directives and regulations where the CE mark is required.
The Single Market was established in 1993 and is still considered one of the most significant achievements of the European Union. The main goal was to ensure the movement of goods and services freely within all the member states and to establish high safety standards for consumers. The CE mark indicates that goods and services do not need to be verified when shipping into another member country. To further support this movement, in April 2011, the Single Market Act was established to boost growth and strengthen confidence in the economy even further.
CE marking is required for many types of products, not just medical devices. The CE symbol can be found on bicycle helmets, toys, laptop batteries, wheelchairs, construction equipment, gas appliances and cell phone chargers - to name a few. CE marking is required for products manufactured anywhere that are sold in the EU, and only for those products for which EU specifications exist and require CE marking. The CE marking signifies that the product has been found to meet the general safety and performance requirements (GSPRs) of the European health, safety and environmental protection legislation and allows the product to be sold in the EU.
Manufacturer responsibilities for CE marking
Medical device manufacturers are responsible for properly and legally CE marking products before they leave the warehouse.
Most Class II and III medical devices, along with IVDs and some Class I devices, require a conformity assessment performed by a Notified Body to ensure that all legislative requirements are met before it can be placed on the market. Manufacturers of most Class I devices can self assess conformity. This process needs to demonstrate that all the legislative requirements are met, including any testing and inspections, and that all necessary certifications are obtained.
The European Commission lists 6 steps that manufactures should follow to affix a CE marking to their devices:
- Identify the applicable directive(s) and harmonized standards - see EU standards for Medical Devices, In Vitro Diagnostic (IVD) devices, and Implantable Medical Devices.
- Verify product specific requirements using the essential principles identified in the above standards.
- Identify whether an independent conformity assessment by a Notified Body is necessary. Notified bodies will be required to verify compliance with relevant Essential Requirements for most medical devices classified as IIa, IIb, or III - along with sterile class I devices. See the Notified and Designated Organization (NANDO) database for available notified bodies.
- Test the product and check its conformity.
- Create and keep available the required technical documentation.
- Affix the CE marking and create the EU Declaration of Conformity.
Importer responsibilities for CE marking
If you are importing medical devices into the EU, it is your responsibility to review all the technical documentation and maintain a copy, or to make sure that it’s available to you upon request.
You should verify:
- That the device has been CE marked and that the EU declaration of conformity has been completed.
- That the manufacturer has designated and established an authorized representative.
- That the device is labeled appropriately and contains instructions for use (IFU).
- When applicable, that a UDI has been assigned to the product.
- Whether or not the product is registered in EUDAMED (registration is currently voluntary).
Take action:
- List your name and address on the device or packaging, in addition to the manufacturer’s information.
- Keep records of complaints, non-conformities, recalls, etc. on file.
- Report any noticed non-conformity or product complaints from end users to the manufacturer and authorized representative immediately.
- Maintain a copy of the EU declaration of conformity and any other relevant certificates.
Distributor responsibilities for CE marking
If you are a distributor, you are responsible for reviewing the technical documentation provided to you so that you can verify the product is safe to put on the local market. You must also be sure the product is labeled correctly with the CE marking symbol clearly visible. The technical file documentation contains all of the information that is necessary to show conformity of the product to the applicable requirements.
You should verify:
- That the device has been CE marked and that the EU declaration of conformity has been completed.
- That the device includes all the appropriate labeling, including instructions for use.
- That if imported, the importer has complied with all the EU regulations.
- When applicable, that a UDI has been assigned to the product.
Take action:
- Report any noticed non-conformity to the manufacturer, importer, and authorized representative immediately.
- If a product appears to be out of compliance to the regulations and could pose a serious risk, the information should be reported to the Competent Authority, and to the manufacturer, importer and authorized representative.
- Any complaints or reports from end users about the product should be reported to the manufacturer and, if necessary, to the importer and authorized representative.
Important note: If the importer or distributor markets the product under their own company name, then they become responsible for CE marketing, and take over that role from the manufacturer.
CE marking is mandatory when importing products into the European Union, which is part of the larger European Economic Area (EEA). The EEA Agreement, established in 1992 and made official in 1994, is an international agreement that enables the extension of the European Union’s single market to non-EU members. It consists of the 27 EU countries plus the four European Free Trade Association (EFTA) countries - Iceland, Liechtenstein, Norway and Switzerland. Today, the EFTA has 29 Free Trade Agreements (FTAs) with 40 countries and territories outside the EU. Because these countries operate in the single market, this allows free movement of goods and services across all of the EEA.

Source: European Environment Agency (EEA).
All medical devices sold in the EU require a CE mark. While a CE mark is not required for items such as chemicals and pharmaceuticals, it can be required for combination devices and medical device software. For these two situations, how do you know if your product requires a CE mark?
To continue reading this ebook, including an overview of CE mark costs, and the associated technical documentation/general safety and performance requirements (GSPRs) that manufacturers are required to maintain please register to download the full version


Your regulatory team needs dedicated regulatory software
Prioritize the needs of your regulatory team
Regulatory affairs teams are responsible for highly critical components of a medtech company’s success, including:
- Market Entry: Meeting go-to-market goals by ensuring accurate, timely market registrations and approvals.
- Regulatory Integrity: Protecting a product’s market position by maintaining regulatory compliance within an ever-changing regulatory landscape.
- Post-market Compliance: Ensuring compliance with post-market surveillance and reporting requirements specific to each market and each device classification.
Your regulatory team should be focused on positioning your products for success in every market you enter. But, did you know that regulatory professionals spend between 30% and 50% of their time looking for the data that they need to do their jobs? This is because many regulatory teams are still using spreadsheets or manually pulling together data from eQMS, PLM, ERP, and other disjointed systems to track the information they need to ensure compliance across markets.
RIM systems compared to eQMS systems
Regulatory Information Management (RIM) systems provide a platform purpose-built for regulatory teams. There are quality systems that provide some regulatory functionality, such as tracking product registrations and even supporting the creation of submission documents. However, these systems fall far-short of providing holistic regulatory management functionality. While functionality can vary among eQMS and RIM systems, RIM systems tend to offer the following features above and beyond what you will find in eQMS modules:
Standards management
International standards play a critical role in regulatory affairs. Regulatory teams need to track which standards are relevant to their products, and link them to product records and/or technical documentation. Any changes to standards can be captured, and impacts to products can be identified automatically. A good RIM system will flag regulatory changes, identify the affected products, and track updates and approvals as needed.
Essential principles management
As more countries and regions around the world adopt Essential principles or GSPR requirements, the regulatory burden of creating and maintaining complex technical files grows. In addition to tracking international standards, RIM systems can fully digitize essential principles/GSPR tables, and allow users to link them to relevant standards and products. This structure allows regulatory teams to easily author, and, more importantly, make automatic bulk updates to tables when standards or product details change.
UDI management
Another growing regulatory burden is the proliferation of UDI requirements. UDI has traditionally been considered a labeling or supply chain concern, but the growing number of country requirements mean that UDI data management is a key component of getting any product to market. RIM systems can manage country-specific UDI data alongside products, registrations, certificates, and other regulatory documents, allowing regulatory teams to ensure that UDI information is correct and up to date.
Regulatory Intelligence
One of the reasons that regulatory teams spend so much time looking for information is that it’s simply hard to find trustworthy regulatory intelligence. Some RIM systems provide a regulatory intelligence database along with their digitization and automation capabilities. This means that regulatory teams have access to quality (well-translated) information, and can use it directly within their processes.
RIM Integrations
RIM systems are also built to integrate with eQMS, PLM, and ERP systems providing a strong regulatory framework across the company, including:
- The ability to use product ‘selling status’ in the RIM system to control the ability to market and sell the product in the company’s ERP system (on a product-by-product and country-by-country basis).
- Ensuring consistent and up-to-date SKU lists between product submissions being created in RIM systems and product development being tracked in PLM systems.
- Triggering regulatory impact assessments when quality records are updated in the eQMS .
- Providing consistency in regulated data among manufacturing, engineering, and regulatory data.
Why you want best-of-breed software: The “all-in-one” myth
Much of the software developed today is designed with integration in mind, and API integrations make it possible to create direct, customized links between separate systems. The days when system integrations meant custom code, batch imports, and clumsy (sometimes unreliable) data synchronization are gone.
This means that organizations can, and should, choose the right software for each task or team. The ability of your regulatory team to work effectively and efficiently is critical to the success of your product market launches, regulatory submissions, and the on-going management of each product in each market. Don’t make them settle for functionality in another team’s system - give them software designed specifically for their needs!
“Best-of-breed,” purpose-built software for each team not only gives everyone in your organization the tools they need to be most productive and successful, but minimizes the costs and risks associated with customizing systems to do something they were not meant to do.
To learn more about RIM tools, read the Rimsys Benefits data sheet.

21 CFR Part 11 for regulatory affairs teams
What is 21 CFR Part 11?
21 CFR Part 11 refers to the federal regulation that address electronic records and electronic signatures associated with FDA requirements. This single, relatively small, part of the Code of Federal Regulations is extremely significant for companies with FDA-regulated products because it impacts every document signature, electronic file, and FDA submission. Codified in 1997, interpretations of this FDA-issued regulation continue to be debated and re-evaluated as the technology supporting electronic records and signatures changes. In this article, we’ll discuss the regulation and generally accepted interpretations.
Note that discussions and statements in this document are our observations only and should not be taken as fact. You can refer directly to the regulation here.
Part 11: General Provisions
The General Provisions section of 21CFR11 addresses the scope of the regulation, when and how it should be implemented, and defines some of the key terms used. It states that the purpose of Part 11 is to define the criteria under which electronic records, electronic signatures, and handwritten signatures attached to electronic records are equivalent to, and as reliable as, handwritten signatures on paper documents.
Fundamentally, any record that is maintained, used, or submitted under any FDA records regulation is subject to Part 11, and the FDA will accept electronic records in lieu of paper records if an organization can prove that their records and systems meet the Part 11 requirements.
The General Provisions subpart also sets forth a number of definitions, and we’ve listed the ones that are most significant to our discussion here:
- Closed System: A computer system or software whose access is controlled by the same people who are responsible for the information stored in the system. Because the opposite of a closed system, and “open system,” is subject to additional scrutiny be sure that you are able to thoroughly explain and provide documentation for a decision to classify your system as a “closed system.”
- Open System: A computer system or software whose access is not controlled by the same people who are responsible for the information stored in the system.
- Digital Signature: An electronic signature created in a manner that can be verified, ensures the identity of the signer, and maintains the integrity of the document and signature. This often involves the use of cryptography and/or biometric data.
- Electronic Signature: Symbols that represent a legally binding equivalent to an individual’s handwritten signature (as adopted and authorized by the signer).
Part 11: Electronic Records
The Electronic Records section sets forth the requirements for administration of closed and open electronic record-keeping systems, then discusses signature manifestations and requirements for establishing a link between signatures and records.
Part 11 defines a “closed system” as any computer system in which the users controlling access to the system are the same people who are responsible for the data in the system. Today, most systems can be classified as closed systems, but take special care to document control procedures around software that is hosted offsite or classified as a SaaS solution.
This section of the regulation deals with the controls that need to be in place for all applicable electronic record systems by defining:
- Procedures to ensure that all electronic records are authentic, have integrity, and can ensure confidentiality (where that is appropriate).
- Validation requirements for systems that maintain electronic records to ensure that all records are accurate, reliable, and that the system performs consistently according to regulatory requirements.
- Audit trail requirements for all regulated records to ensure a complete history of all changes to records are maintained.
- Controls around system access and document signatures.
Part 11: Electronic Signatures
The Electronic Signatures section defines the components of electronic signatures and the required controls and procedures necessary for using them.
In general, an organization must be able to demonstrate that electronic signatures:
- Are unique to each individual, and that the individual assigned an electronic signature has had their identity and level of authorization verified.
- Must be based either on biometric data (such as fingerprints) or made up of two distinct pieces (ie: a User ID and password)
- Require appropriate controls to ensure that they are verified periodically, cannot be used by someone other than the intended user, and are immediately deactivated if compromised in any way.
Practical application of 21CFR Part 11 for regulatory affairs professionals
21 CFR Part 11 is a critical regulation, and one that can be open to interpretation. Below, we cover some of the key areas that should be of concern for RA professionals. This is an overview of key areas only, and should not be taken as complete instruction or guidance for 21CFR part 11 compliance.
System compliance and validation
Any system that you are using to store electronic records that fall under FDA regulations needs to be compliant with Part 11. This includes everything from spreadsheets to full-featured RIM and document management systems.
Software vendors will often document how their systems are developed to be compliant, and may even support system validation during implementation - but it is ultimately the responsibility of the user organization to ensure that their systems and processes are compliant with Part 11. System validation is the process of documenting that your system meets all of the Part 11 requirements. Software vendors can support this process by ensuring that their systems are built on a highly secured infrastructure that can be demonstrated and proven.
The Rimsys system was built from the ground up to meet the stringent requirements of not only 21 CFR Part 11, but other industry standards and good practices guidelines (GxP). We have put in place a rigorous validation program, built by industry experts and supported by a secure and well-documented infrastructure. For more information, visit the Rimsys Security and Privacy page.
Audit trails
Audit trails are the required system logs that track the who, when, and what of every change made to data that falls under Part 11. Audit trails should be generated and time-stamped by the system, with no ability for users to change that information. Audit trails serve two purposes under 21 CFR Part 11:
- To demonstrate that documented policies and procedures are being followed, including that only users with the appropriate authority are managing data.
- To prove that data retention policies are being adhered to (see below).
At any time, you should be able to view the history of any record, from a Design History File to a submission document, in order to determine what changes have been made, when they were made, and by whom.
Record retention
21 CFR Part 11 specifies that electronic records must be protected and readily available throughout the defined record retention period. Additionally, 21 CFR Part 820 specifies that records related to the quality, manufacturer, regulatory submissions, or any other data that falls under FDA regulation, should be maintained for the life of the medical device and for a minimum of two years from the date of first commercial distribution. This is often referred to as “cradle to grave” tracking.
This means that regulatory professionals need to not only be aware of their company’s record retention policy, but need to ensure that any system being used to track regulatory submissions or other data subject to audit meets Part 11 and Part 820 requirements. Note that record retention requirements apply also to paper records where they are the source document.
Electronic and digital signatures
An important piece of 21 CFR Part 11 is its definition of electronic and digital signatures. “Electronic signature” is used to define any set of symbols that are used in place of a handwritten signature, whereas a “digital signature” is an electronic signature based on methods that ensure the identity of the signer where the integrity of the data can be verified. A digital signature can be based on biometric data (such as fingerprints) or secure user IDs and passwords that are controlled to ensure only one authorized user can use the signature.
As a regulatory affairs professional, you should ensure that:
- Everyone on your team who needs to sign documents has their own unique digital signature and understands the importance of protecting it. Sharing of electronic credentials is a common FDA audit observation. Also ensure that users who are not required to sign documents have appropriate access to data to discourage other users from sharing login credentials with them.
- You are following your company’s policies concerning electronic signature audits so that passwords remain updated and strong and signatures are revoked when a user leaves or changes positions.
- You immediately report any possible loss, theft, or sharing of user credentials or devices that generate identification codes.
While 21 CFR Part 11 is usually considered more of a “quality regulation,” it is important that regulatory teams within medical device organizations fully understand this regulation and its compliance implications. To learn more about the regulations, click below to read our regulatory brief.

The importance of PLM, eQMS, and RIM systems for medical device manufacturers
Medical device manufacturers around the world are facing an ever changing and increasingly demanding regulatory environment. This growing complexity is driving a renewed focus on digital transformation within the medtech industry, leading companies to reevaluate, expand, and update current systems. Ensuring that your company has the right software in place to implement strong processes and controls around product development, product quality, and regulatory compliance is critical. Relying on an eQMS, PLM, or RIM system that isn’t purpose-built for your needs is likely to provide inconsistent levels of functionality across your organization, and also lead to potential compliance issues.
For example, an ERP system with a configure-to-order or strong engineering focus may provide a core PLM functionality, but only a small quality module that was added late to the product. In this case, your quality and regulatory teams could be left to build custom functionality or work in spreadsheets outside of the system. To ensure that everyone in your company has the functionality they need, consider best-in-breed software for each team - including the engineering and product development, quality, and regulatory teams. Today’s technology is built with integration in mind, and there are strong reasons for integrating your PLM, eQMS, and RIM systems.
In this article, we will provide an overview of PLM, eQMS, and RIM systems - their core capabilities, strengths, and what to consider if your company is a medical device manufacturer looking to add or update software systems.
PLM for medical device manufacturers
Product Lifecycle Management (PLM) systems are typically used by product development and engineering teams to optimize resources, shorten product development time, and manage a product throughout all phases of its life. The primary functions of a PLM system include:
- Change management (ECN and ECO control)
- Product history and revision management
- Configuration management
A PLM system for a medical device manufacturer is used to mange each product’s design history file (DHF) and device master record (DMR). The PLM system will store bills of material for each revision of the product, and can therefore be integrated to a core ERP system to ensure that production adheres to the approved design.
PLM systems manage the workflow around product changes, typically including both Engineering Change Notices (ECN) and Engineering Change Orders (ECO). Change requests and execution, including reviews and approvals, can therefore be managed through one system. Medical device manufacturers may have issues, however, tracking product changes not related to the device components themselves, such as labeling changes. While it is certainly possible to track non-product changes within the BOMs of a product in PLM or ERP systems, many medical device manufacturers may move the ECN process to their eQMS system, and manage product-based ECO’s in the PLM system.
eQMS for medical device manufacturers
Quality Management Systems are built around strictly controlled workflows and closed-loop processes. Unlike a PLM system, an eQMS (Enterprise Quality Management System) system is not product-focused, it is process-focused. Some of the items that an eQMS provides centralized control for, include:
- Document control
- Non-conformance tracking
- CAPA management
- Risk management
- Supplier quality control
A strong eQMS system allows medical device manufacturers to establish quality controls from supplier to customer, and is critical for meeting the requirements of 21CFR Part 11, 21CFR Part 820, and other quality and electronic records regulations.
According to Qualio, a leading provider of eQMS systems for the life sciences industry, there are 5 Indispensable Features of Enterprise Quality Management Software:
- Company-specific features unique to your requirements - that align with the needs of your team and your processes, so that you don’t need to spend significant effort customizing and configuring the system.
- Ability to integrate processes - in order to integrate with data and processes from other systems (such as PLM and RIM systems).
- Flexible and expandable - allowing the system to grow with your company as you need new features and functionality.
- In-depth reporting capabilities - to give your teams visibility into the data they need to make effective decisions every day.
- Meets compliance standards - to make audits and compliance as easy to manage as possible.
RIM systems for medical device manufacturers
Regulatory Information Management (RIM) systems facilitate and automate regulatory activities. Regulatory affairs professionals are responsible for managing increasingly complex regulatory requirements, often across many markets. This means that RA professionals spend more than 50% of their time looking for data needed to complete regulatory submissions and ensure compliance with updated regulations. RIM systems centralize, organize, and manage all regulatory information, while automating and streamlining the regulatory processes around it. It is also worth noting that the first RIM systems were designed for the pharmaceutical industry, and did not meet the needs of medical device RA teams. RIM systems specific to the medical device industry have more recently come to market to address the needs of medtech RA teams and the increasingly complex devices and regulatory landscape they work with.
The key capabilities of a medical device RIM system:
- Product and registration management is the most central piece of functionality in any RIM system. RIM systems are product-focused (as are almost all RA activities) and enable detailed product information to be centralized and tracked, including registration and selling status by market.
- Regulatory submissions are an important and time-consuming responsibility for RA professionals. RIM systems can provide country-specific submission templates, integrate to product and quality information in PLM and eQMS systems, and allow you to manage the workflow around creating a submission document - not to mention the assembly of the final submission package.
- Regulatory intelligence is provided in some RIM systems, and solves a major challenge faced by RA teams. Regulatory requirements not only differ across markets, but can change frequently. A RIM system with a regulatory intelligence component delivers up-to-date, market-specific regulation information, along with monitoring to alert your RA team of changes.
- Essential principles and standards management supports the creation and maintenance of technical files, and GSPR/Essential principles checklists. This significantly reduces the time it takes RA teams to document when standards or product details change.
- UDI and label management may be handled separately from other regulatory activities in your organization, but integrating them within a single RIM system can simplify data collection and management, and electronic submissions to government databases.
- Project management capabilities are important in any RIM system, enabling the management of tasks, requests, and approvals around regulatory activities. RIM systems provide the traceability that regulatory teams need by keeping a detailed history of every project task, update, and approval.
- Reports and dashboards available in a RIM system provide RA teams with the information they need to understand how long regulatory submission and other processes typically take, product and market-specific registration information, and other insights that pull from the large amount of data stored in your RIM system, allowing your RA team to function as efficiently as possible!
Do medical device manufacturers need PLM, eQMS, and RIM?
We might be a bit biased, but we feel strongly that the answer to this question is “Yes.” Why? Because product development teams are expected to release new products more quickly than ever, quality teams need to ensure company-wide process meet multiple quality standards, and regulatory teams are facing an increasingly complex and quickly changing regulatory landscape. Each of these teams needs functionality built specifically for them to ensure that they are as efficient and effective as possible. Delaying a product launch, failing a quality inspection, or missing a regulatory submission deadline are not acceptable outcomes. Combine this with the fact that today’s systems are truly built with integration in mind - so that information can be shared, not duplicated, across systems. Learn more about the system that Rimsys integrates with.
If you are interested in learning more about RIM systems - read our RIM Buyer’s Guide for MedTech Companies.
And if you are interested in learning more about the Rimsys RIM system - schedule a personalized demo to see the product for yourself!

Software as a medical device (SAMD) - classification overview
What is software as a medical device (SaMD)?
As the pace of technological innovation continues to increase, the definition of what constitutes a medical device also continues to evolve as countries update regulations. In 2013, the International Medical Device Regulators Forum (IMDRF) created the Software as a Medical Device working group. Currently chaired by the U.S. FDA, the working group is chartered with developing guidance that encourages innovation while assuring safe and effective products. While SaMD is regulated differently in different countries, this article will focus on the many similarities, and some differences, between the FDA regulations in the U.S. and the MDR regulations in the EU.
In general, medical device software falls into 3 different categories:
- Software as a Medical Device (SaMD): The IMDRF defines SaMD as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” We list specific examples below, but typically the software classified as SaMD isdesigned to run on generally available hardware, such as Windows computers or mobile devices, or online in the “cloud”. While they may be utilizing data from another medical device, SaMD performs its function independently of any medical equipment or hardware.
- Software in a Medical Device: Sometimes referred to as SiMD, Software in a medical device cannot operate separately from its device, or perform its primary function without the device. For example, the software used to program and run an MRI machine would be useless without the MRI machine. Software in this category is regulated together, and as part of, the whole device.
- Software as an Accessory to a Medical Device: Similarly to Software in a Medical Device, software in this category cannot fulfill a medical purpose on their own. In some cases, a manufacturer may be able to bundle or embed the software in the medical device (making it SiMD) and/or also sell the software separately, making it an accessory.
Similarly, in Europe, the European Commission’s Medical Device Group (MDCG) defines Medical Device Software (MDSW) as a having its own intended purpose. Software which controls a medical device or is otherwise part of a medical device and does not serve a separate medical purpose does not qualify as MDSW, but is regulated by the MDR.
Software in the SaMD category is both a medical device AND software - with relevant regulatory and quality considerations that are specific and unique to each category, yet which must work in tandem. For example, software development best-practices, referenced by the IMDRF SaMD working group, call for iterative feedback loops allowing for quick turnaround of feature requests and bug fixes. While the ability to provide the latest technology and features to the market is an important advantage with SaMD, it does not supersede applicable medical device regulations governing patient safety and efficacy.
Examples of software as a medical device (SaMD)
Because SaMD software, by definition, is capable of running independently of any specific medical device or hardware, there is a relatively clear line between Software as a Medical Device and other medical software:
SaMD regulatory overview and framework
Why does SaMD have independent regulatory considerations?
Over the past decade, the IMDRF, FDA, and other regulatory bodies have worked to better align regulations with the quickly evolving capabilities and nature of digital devices. Software-only devices (SaMD) generate unique opportunities and considerations:
Because SaMD software typically runs on publicly available hardware:
- SaMD software must not only be designed to work on specific platforms (usually multiple), but must also be tested and updated frequently as new hardware and operating systems become available.
- Agile software development methodologies can provide an environment in which fast product feedback loops are supported within the required regulatory framework.
Because SaMD software can be made readily available to the general public, or specific patient groups, using their own devices:
- SaMD software can generate faster user/patient feedback - both for medical professionals and for the device manufacturer. Given this environment, regulatory bodies generally want to enable the market to safely take advantage of new innovations as quickly as possible.
- Product testing needs to take into account the unique and varied environments in which the software may be used, potentially in environments the product is not intended for. The software developer cannot control updates to operating systems or internet browsers, other software that may be running on a user’s device, or the ability for the user to potentially share software or data with others.
- The advantages of quickly delivering product fixes and updates need to be measured against potentially introducing new, possibly misunderstood or unwanted features, to all users at once.
SaMD and MDSW risk-based categories
As with all medical devices, the FDA and European MDR classify SaMD and MDSW, respectively, based on the potential impact to patient or public health. The SaMD categorization framework from the IMDRF is an effort “to introduce a foundational approach, harmonized vocabulary, and general and specific considerations, for manufacturers, regulators, and users alike to address the unique challenges associated with the use of SaMD...” This framework provides guidance only for today’s medical software developers, as well as regulatory bodies, such as the FDA. The chart below lists these risk categories by the state of the healthcare situation or condition and by the significance of the information provided by the SAMD:
SaMD categories
SaMD Category I:
SaMD Category I software can provide information for both Serious and Non-Serious health conditions or diseases. Software dealing with serious conditions can be classified in Category I only if it is providing information to inform, not drive, clinical management and is of low impact. Otherwise, Category I SaMD software provides information related to non-serious diseases or health conditions.
Example: Software that collects exercise-related data, such as heart rate, number of steps, and distance walked. If the information is stored and/or transmitted for use by a qualified professional the software is considered to be in Category I, as long as the information is not used by the software to make treatment recommendations directly to the patient or healthcare provider.
SaMD Category II:
SaMD software in Category II may be used to provide information relevant to a non-serious, serious, or critical healthcare condition or disease, depending on the significance of the information it provides to the healthcare decision. Software that is used to treat or diagnose a health condition is only classified in Category II for non-serious health conditions. Software that provides information regarding serious or critical health conditions or diseases will be classified in Category II only if it provides information to drive or inform, respectively, clinical management decisions (as opposed to providing treatment or diagnosis recommendations directly).
Example: SaMD that monitors a diabetic patient’s carbohydrate intake and blood glucose level to calculate recommended insulin dose.
SaMD Category III:
SaMD software that provides information to treat or diagnose serious conditions - or which drives clinical management for a critical condition - is classified in Category III.
Example: SaMD that analyzes individual data from at-risk populations for a specific type of cancer, and is used to develop preventative intervention strategies.
SaMD Category IV:
SaMD Category IV software provides information used to treat or diagnose a critical health condition and is considered to be very high impact.
Example: SaMD that evaluates images of skin lesions in order to determine malignancy.
For SaMD intended to be used in multiple healthcare situations, the software will be categorized at the highest category according to the SaMD definition statement.
SaMD within the FDA and MDR regulatory framework
The graph below relates SaMD categories with FDA medical device classes, and further identifies where an independent review is recommended by the FDA. For additional information see Software as a Medical Device (SaMD): Clinical Evaluation. Guidance for Industry and Food and Drug Administration Staff.
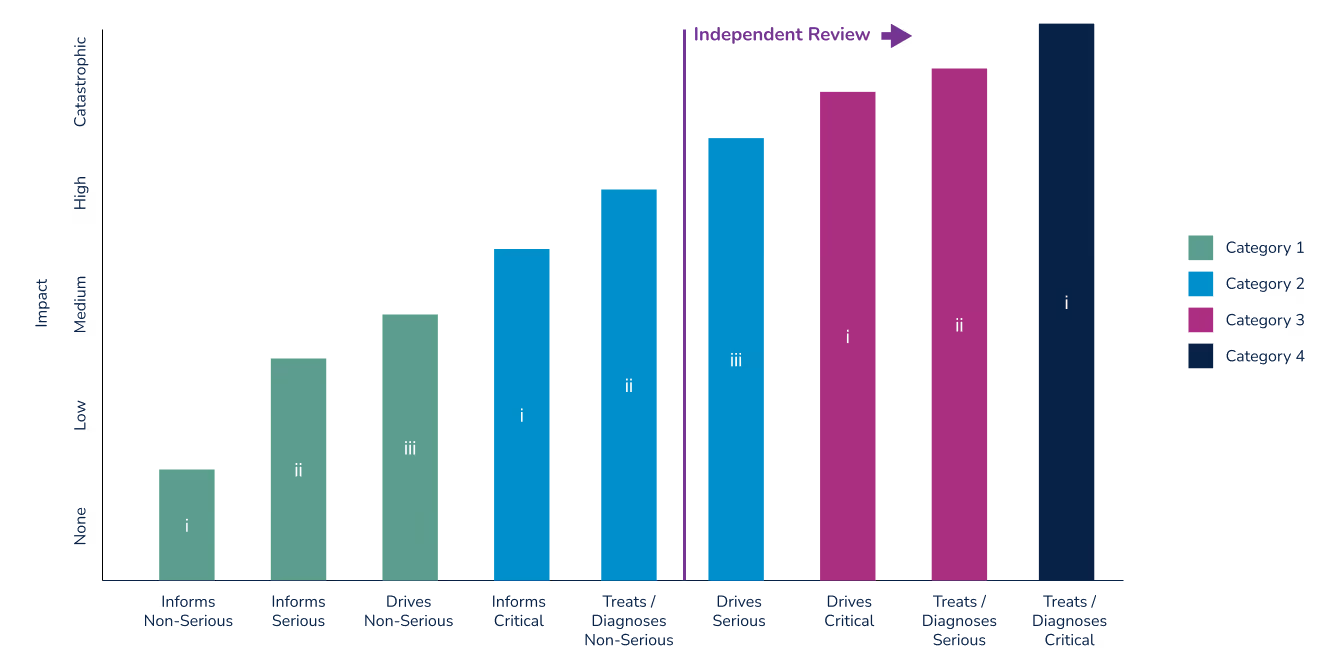
Note that FDA regulatory classifications more closely align with the IMDF’s SaMD categories than do those defined by under the European MDR. In the U.S., a device is classified by identifying similar, predicate devices. The simpler 510(k) pre-market submission can be used if a manufacturer can show that their device is substantially equivalent to a Class I or II device. With few exceptions, devices that are Class III are subject to the more rigorous Pre-Market Authorization (PMA) process. Devices where no substantial equivalent can be shown are subject to to the PMA or De Novo regulatory submissions processes. The De Novo process was implemented in 2010 to provide a pathway for novel devices with lower risk profiles.
Medical Device Software is classified by the European Commission’s Medical Device Coordination Group (MDCG) into Class I, II, or III. For devices falling under the IVDR, the MDR defines four classes using letters; A, B, C, and D.
In the EU, a rules-based framework is used to classify devices. MDSW classification is relatively complex, using a series of 22 “waterfalling” rules with yes/no questions that lead to a final classification of each device. While we won’t go into detail in this article on each of the rules, Rule 11 does deserve discussion. (You can read the Guidance on Classification of Software for MDR and IVDR here).
The MDR’s “rule 11” regarding MDSW classification was released in 2017. This rule states, in part, that “Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa.” The rule then provides 2 exceptions that increase the MDSW classification in cases where the decisions could cause “a serious deterioration of a person’s state of health or a surgical intervention” (class IIb), or could cause “death or an irreversible deterioration of a person’s state of health” (class III).
As a result of this rule, very few MDSW products can be classified as Class I, meaning that the majority of MDSW is subject to conformity assessments by a Notified Body. Medical software manufacturers marketing their product in Europe should therefore not rely on previous product classifications as a guide.
The future of regulatory approval for software as a medical device (SaMD)
The FDA recently released a new draft guidance document addressing the Content of Premarket Submissions for Device Software Functions. This is a major update of the 2005 guidance on pre-market submissions for software in medical devices and is also long overdue, given the pace of change in the software industry (the FDA originally committed to delivering this update in their fiscal year 2019 as part of the MDUFA IV agreement). This guidance sets out requirements for all pre-market submissions, including 510(k), DeNovo, and PMA submissions.
The recent draft document provides additional guidance around the documentation required for premarket submissions. It defines an “enhanced” level of documentation required for software that is a Class III device, combination product, Blood Establishment Computer software, or when the failure of the software would present probable risk of death or serious injury.
Staying on top of medical device classifications
Classifying a medical device and gaining regulatory clearance can be a complex process, especially if the device contains software or other emerging technology where some regulatory bodies may be further along than others in developing applicable regulations. For additional information about some of the common pathways to market for new products in the U.S., read our Beginner’s Guide to the FDA 510(k) and Beginner’s Guide to the FDA De Novo Classification Process.
Interested in learning how Rimsys can automate your submission process? Request a custom demo of our RIM platform.

PSUR: Periodic safety update reports for medical device and in vitro diagnostic products
For many medtech companies, structured, post-market surveillance reporting requirements are a new component of a product’s regulatory lifecycle. The EU MDR/IVDR regulations introduced a host of new post-market surveillance requirements for medical devices and in vitro diagnostics made available for sale in the EU—including regular summary reporting to be recorded within the device technical file or submitted directly to Notified Bodies. This article provides a brief background of Periodic Safety Update Reports (PSUR), the types of products which they’re applicable to, and what content is typically included in a PSUR.
What is a PSUR?
The Periodic Safety Update Report or PSUR is not a new term, at least to the pharmaceutical community. The industry has been operating with regulations related to PSUR for some time. But for the medical device and IVD community, it’s a new requirement that stems from EU Regulations MDR 2017/745 (article 86) and IVDR 2017/746 (article 82). A PSUR is basically a report summarizing critical actions and conclusions derived from post-market surveillance data of a medical or in vitro diagnostic device. All associated preventive and corrective actions should be documented throughout the lifetime of the device, even if the product is no longer on the market.
The introduction of the PSUR under the MDR and IVDR requires a more consistent, standardized, and systematic review of all Post Market Surveillance (PMS) data by medical and IVD device manufacturers. The PSUR is meant to summarize the results and conclusions of the analysis of the post-market surveillance data that has been gathered, resulting from the activities detailed in either the Post-Market Surveillance Plan (PMSP). In addition, any rationale and description of any preventive and corrective actions taken for safety reasons should be included.
The PSUR is for specific classes of medical and IVD devices, as per the table below:
Note: a European competent authority or Notified Body can request your PMSR or PSUR at any given time.
What is the purpose of a PSUR?
The purpose of a PSUR is for manufacturers to demonstrate with objective evidence that they have designed and deployed a Post-Market Surveillance system which uses data to drive action within their Quality Management System and ensure the continued safety, performance, and efficacy of their devices. It’s intended for moderate and high-risk devices (MD Class IIa, IIb, III: IVD Class C and D) and provides a detailed summary of results and conclusions derived from the PMS data.
What’s included in a PSUR?
Medical device and IVD manufacturers need to prepare a PSUR for each device, and where relevant, for each category or group of devices. The manufacturer is responsible for preparing and updating the PSURs and making it part of the technical documentation that should be included with the Essential Principles/GSPR’s. These reports must be clear, organized, searchable and in easy-to-read format.
The PSUR should be a stand-alone document. While the content of a PSUR can vary, depending on the amount of specific data the vendor chooses to include, the PSUR should, at a minimum, always include: an executive summary, safety conclusions and benefit-risk determination, main findings of the Post Market Clinical Follow-up (PMCF) [or Post Market Performance Follow-up for IVDs], vigilance data, information about sales volume, user population, and usage frequency. A PSUR is meant to provide an overview of information, not to be a complete duplicate of all the PMS report information.
Something very important to note, A PSUR is required throughout the lifetime of the device plus the shelf-life where relevant. So for example, A single use device could have a lifetime of 1 year, but a shelf life of 5 years. After the end of device production, the PSUR can be stopped only when the cumulative data of the PSUR issued for this device covers the duration of the shelf life (6 years).
What is the format of a PSUR?
The PSUR format is composed of two elements: the PSUR form and the PSUR report.
The PSUR report is a PDF file that the manufacturer will be required to upload in EUDAMED for class III devices and for implantable devices. The PSUR form is an electronic form that will be completed by the manufacturer in EUDAMED, after they have finished the “completeness” check.
The PSUR form contains all your relevant administrative information as well as data to identify and distinguish between different PSURs for the same device. It should also contain data necessary for the registration of the PSUR in EUDAMED. The PSUR form will be available by the Commission on their website at a later date to be announced.
The PSUR report will contain all of the core content including the executive summary, grouping of devices, sales volume, and PMS data discussed in the previous section.
Keeping on top of technical documentation
PSUR requirements, and PMR data are now a critical part of the technical documentation that regulatory affairs professionals in medtech are required to maintain. Along with the expanded GSPR requirements that come with the MDR/IVDR rollouts, traditional approaches to managing technical docs are no longer effective, and can be prohibitively time-consuming to maintain. Regulatory Intelligence Management (RIM) systems, like Rimsys, can provide a much more powerful, effective, and streamlined way to manage all of a products’ technical files and supporting documentation.
To learn more about RIM systems, read our case study to see how a global leader in in vitro diagnostics was able to reduce the time spent on maintaining technical docs by 99% or request a custom demo of the Rimsys platform.

De Novo classification process: a beginner's guide
This article is an excerpt from The beginner's guide to the FDA De Novo classification process ebook.
Contents
- Introduction
- Chapter 1: What is an FDA De Novo request?
- Chapter 2: Contents of a De Novo request
- Chapter 3: Submitting a De Novo request
- Appendix A: Acceptance review checklist
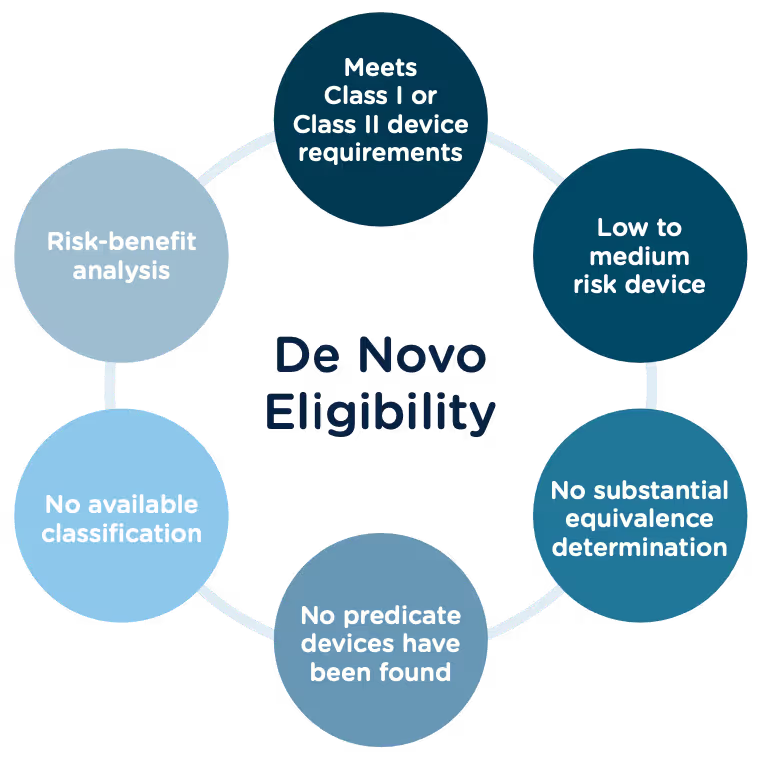
Congratulations, you have successfully developed a new medical device! Now you need to take it to market. Normally in the United States this would mean completing a 510(k) submission. However, the 510(k) relies on “substantial equivalence”—a comparison to a similar device already on the market (also called a predicate device) to assess the risk profile of the new device. What if your device is totally new, and there isn’t a similar device to compare it to? Enter the FDA De Novo process. The De Novo process provides a pathway to market for novel devices with a low to medium risk profile.
What does De Novo mean?
According to the Merriman-Webster dictionary, de novo is a Latin word meaning “as if for the first time; or anew.” Perfectly fitting that the FDA uses this term “De Novo” to describe market approval requests for new medical devices or technology where there is no comparable predicate device on the market.
The Food and Drug Administration Modernization Act of 1996 provided the FDA with the authority to create the De Novo Classification Process. It's a process that uses a risk-based strategy for a new, novel kind of medical device, in vitro diagnostic, or medical software solution whose type has previously not been identified and/or classified. It’s a process by which a novel medical device can be classified as a Class I or Class II device, instead of being automatically classified as Class III, which may not be appropriate. Before the implementation of the De Novo process in 1997, all the “not substantially equivalent” (NSE) products were required to be initially classified as a Class III device. But for a lot of devices, this risk class didn’t really make sense. The De Novo process provides a pathway for more accurate classifications of novel, lower-risk devices.
October, 2021, the FDA released a final guidance document "De Novo Classification Process (Evaluation of Automatic Class III Designation)" to provide guidance to the requester (also known as the manufacturer) and the FDA on the process for the submission and review of a De Novo Classification Request under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act). This process provides a pathway to an initial Class I or Class II risk classification for medical devices for which general controls or general and special controls, provide a reasonable assurance of safety and effectiveness, but for which there is no legally marketed predicate device. This guidance document replaced the "New Section 513(f)(2) – Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff" document, dated February 19, 1998.
Consistent with the final rule, the FDA updated the guidance documents below to provide recommendations for submitting De Novo requests, as well as criteria and procedures for accepting, withdrawing, reviewing, and making decisions on De Novo requests, effective January 3, 2022.
- User Fees and Refunds for De Novo Classification Requests
- FDA and Industry Actions on De Novo Classification Requests: Effect on FDA Review clock and Goals
- Acceptance Review for De Novo Classification Requests
The 510(k) and the De Novo processes are similar in that they are both pathways to market for medical devices with low to moderate risk, which is Class I and Class II. The biggest difference between the two is that the 510(k) heavily relies on the concept of "substantial equivalence" to an existing medical device. You must prove this to get the clearance of your 510(k) submission. In the De Novo process, there isn’t a product currently on the market that is “substantially equivalent” to yours, so it’s like starting with a clean slate. For more on the 510(k) process, see our Beginner’s Guide to the 510(k) ebook.
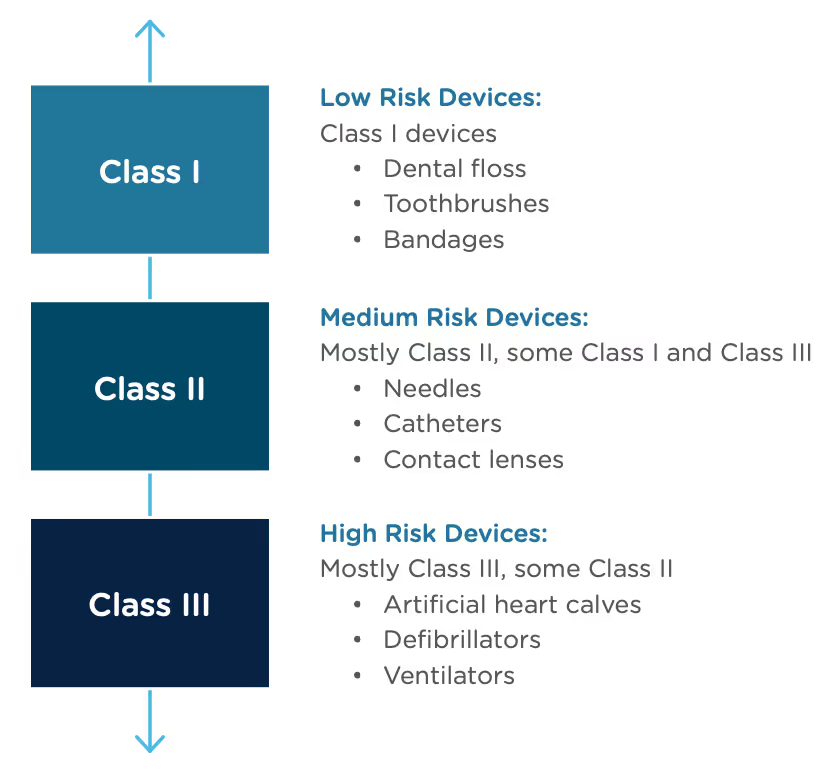
A result of the De Novo process to be aware of is that a successful submission will lead to a new predicate device type that someone else can reference to bring their product to market through the 510(k) process. You’ve done all the work, so now it’s available for anyone to use to provide "substantial equivalence".
De Novo history/timeline
Preparing a De Novo request
1. Do your research! Be sure to complete all the necessary research prior to your submission. You want to be sure that your device is not substantially equivalent to an existing device. Resources to review include:
- The Center for Devices and Radiological Health (CDRH)
- U.S. FDA Device Classification Database
- Device Classification Under Section 513(f)(2)(De Novo)
2. A De Novo request can be submitted with or without a preceding 510(k). There are two options for when you can submit a De Novo request:
Option A: After receiving a not substantially equivalent (NSE) determination (that is, no predicate, new intended use, or different technological characteristics that raise different questions of safety and effectiveness) in response to a 510(k) submission.
Option B: If you’ve determined, after extensive research, that there is no legally marketed device on which to base a determination of substantial equivalence.
3. Be sure all fees are paid to the FDA in advance of submitting a De Novo request. The FDA’s fiscal year begins in October and runs through the following September. Fees have increased each year since they were introduced, but the FDA’s percentage of reviews completed within the 150-day window has increased as well.
A business that is qualified and certified as a “small business” is eligible for a substantial reduction in most of the FDA user fees, including De Novo. The CDRH is responsible for the Small Business Program that determines whether a business is qualified.
Medical Device User Fee Amendments (MDUFA) guidance documents can provide more detailed information about all FDA user fees.
4. The initial request process serves only to determine if the De Novo request is administratively acceptable based upon the Acceptance Checklist. The initial acceptance is followed by substantive review which will determine the final risk classification of your device.
5. A Pre-Submission (Pre-Sub) is a formal written request for feedback from the FDA that is provided in formal written form, and then followed by a meeting. Although a Pre-Sub is not required prior to a De Novo request, it can be extremely helpful to receive early feedback, especially for devices that have not previously been reviewed under a 510(k). If you think you would like to submit a pre-sub first, there are suggested guidelines for submission you should consider:
- Describe your rationale for a Class I or Class II classification for your device.
- Provide the search results of FDA public databases and other resources used to determine that no legally marketed device and no classification for the same device type exists.
- Provide a list of regulations and/or product codes that may be relevant.
- Provide a rationale for why the subject device does not fit within and/or is different from any identified classification regulations, based on available information.
- Identify each health risk associated with the device and the reason for each risk.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed in order to collect the necessary data to establish the device’s risk profile.
- Provide information regarding the safety and effectiveness of the device. Cite the types of valid scientific evidence you anticipate providing in your De Novo request, including types of data/studies relating to the device’s safety and effectiveness.
- Briefly describe any ongoing and/or planned protocols/studies that need to be completed to collect the necessary safety and effectiveness data.
- Provide protocols for non-clinical and clinical studies (if applicable), including how they will address the risks you anticipate and targeted performance levels that will demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.
- Share any proposed mitigation measure(s)/control(s) for each risk, based on the best available information at the time of the submission. Highlight which mitigations are general controls and which are special controls and provide details on each.
- Include any other risks that may be applicable, in addition to those identified in the Pre-Sub, given the indications for use for the device.
- If applicable, provide any controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device.
- Provide any non-clinical study protocols that are sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn. These protocols should address whether the identified level of concern is the appropriate level of concern for the device software, and if any additional biocompatibility and/or sterility testing is required.
- If clinical data is needed, provide information to show that the proposed study design and selected control groups are appropriate?
6. The FDA will attempt to review the De Novo request submission within 15 calendar days of receipt of the request to make a determination that the submission is declined or accepted for review. If they are unable to complete the review within the 15 days, your submission will automatically move to “accepted for review” status. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/de-novo-classification-process-evaluation-automatic-class-iii-designation
7. There are times when the FDA will refund your application fee. They have created a guidance document “User Fees and Refunds for De Novo Classification Requests” for the purpose of identifying:
- the types of De Novo requests subject to user fees
- exceptions to user fees
- the actions that may result in refunds of user fees that have been paid
When is a De Novo request subject to a user fee?
When will the FDA refund a De Novo user fee?
What fee must be paid for a new device submission following a De Novo “decline” determination?
To continue reading this eBook including a detailed walk-through of all the Traditional 510(k) components, submission requirements and timelines, and an overview of the other 510(k) forms including the Abbreviated 510(k) and the Special 510(k), please register to download the full version.
